Fig. 1
Immunostaining for GABA in a semithin (0.5 μm) section from the L4 segment of a rat that had undergone chronic constriction injury of the left sciatic nerve 2 weeks previously. Panel (a) shows a low magnification view through the dorsal horn on both sides, while panels (b) and (c) show higher magnification views of the medial parts of the ipsilateral and contralateral dorsal horns, respectively. Numerous immunoreactive and non-immunoreactive cell bodies can be seen in the dorsal horn on each side, and there is no detectable difference between the two sides. Stereological analysis showed that the proportion of cells in each of laminae I, II and III that were GABA-immunoreactive did not differ between ipsilateral and contralateral sides and did not differ from that seen in naïve or sham-operated animals. Scale bars: a = 200 μm; b, c = 50 μm. Reproduced with permission from Polgár et al. (2003)
Interneurons in lamina II have been studied most extensively and have been divided into four main morphological classes: islet, central, vertical and radial cells (Grudt and Perl 2002; Yasaka et al. 2007). However, even in this lamina, not all cells can be assigned to one of these classes, and the relationship between morphology and neurotransmitter type is not entirely straightforward. A correlative electrophysiological and anatomical study of ~60 lamina II neurons showed that all islet cells (which are defined by their highly elongated rostrocaudal dendritic trees) were inhibitory but that the remaining inhibitory interneurons did not show any characteristic morphological properties (Yasaka et al. 2010).
An alternative approach to classifying the inhibitory interneurons has been to use neurochemical markers. In this way, we have identified four nonoverlapping populations of inhibitory cells in laminae I–III of the rat, based on expression of neuropeptide Y (NPY), galanin, neuronal nitric oxide synthase (nNOS) or parvalbumin (Tiong et al. 2011). Between them, these cells account for over half of the inhibitory interneurons in laminae I–II and a smaller proportion of those in lamina III. The parvalbumin cells correspond to some of the lamina II islet cells (and have similar morphology in lamina III), while the other three neurochemical types are not islet cells and are morphologically heterogeneous (AJ Todd, N Iwagaki and RP Ganley, unpublished data).
There are several mouse lines in which subsets of inhibitory interneurons express green fluorescent protein (GFP), and these have been used to investigate the functions of these cells. These include the GIN mouse (Oliva et al. 2000), in which around one-third of GABAergic neurons in lamina II express GFP; the PrP-GFP mouse (Hantman et al. 2004), in which GFP labels a subset of the nNOS- and galanin-containing GABAergic neurons (Iwagaki et al. 2013); and the GlyT2-EGFP mouse, in which glycinergic neurons are labelled.
1.3.2 Excitatory Interneurons
All of the neurons that are not projection cells and lack GABA and glycine-immunoreactivity are thought to be glutamatergic (excitatory) interneurons, and these account for 60–70 % of the neuronal population in laminae I–III. Initially, it was difficult to be certain of their neurotransmitter phenotype, but this can now be confirmed by their expression of vesicular glutamate transporter 2 (VGLUT2) (Maxwell et al. 2007; Schneider and Walker 2007; Yasaka et al. 2010). Some of these cells belong to the vertical or radial classes, but many cannot be assigned to any morphological class. Although there are several neurochemical markers (e.g. certain neuropeptides, the calcium-binding proteins calbindin and calretinin, protein kinase Cγ) that are largely restricted to excitatory interneurons, little is yet known about whether these represent discrete functional populations (Todd 2010).
1.4 Descending Pathways
The main descending projections from the brain are the monoaminergic systems, which originate in the raphe nuclei of the medulla (serotonergic) and the locus coeruleus and adjacent regions of the pons (norepinephrinergic). However, there is also a GABAergic/glycinergic projection from the ventromedial medulla that arborises throughout the dorsal horn (Antal et al. 1996).
1.5 What We Know About Synaptic/Neuronal Circuits in the Dorsal Horn
Although our knowledge concerning the spinal cord circuits that process somatosensory information is still extremely limited, there has been some progress in identifying specific pathways (Fig. 2). It is likely that most (if not all) dorsal horn neurons receive glutamatergic synapses from both primary afferents and excitatory interneurons, as well as GABAergic, glycinergic or mixed synapses from local inhibitory interneurons. However, there is emerging evidence that these are organised in a selective way, with certain types of primary afferent and interneuron preferentially innervating specific types of dorsal horn neuron (Todd 2010). The inhibitory synapses described above are formed by the axons of inhibitory interneurons and the dendrites or cell bodies of other neurons (axodendritic or axosomatic synapses), and these underlie postsynaptic inhibition, which is the major form of inhibition in the dorsal horn. However, some GABAergic axon terminals form synapses onto primary afferent axons (axoaxonic synapses), and these generate presynaptic inhibition, which can control specific types of sensory input. In addition, there are dendrodendritic (and dendroaxonic) synapses, in which the presynaptic element is the dendrite of a local GABAergic neuron. The functions of these synapses are not well understood.
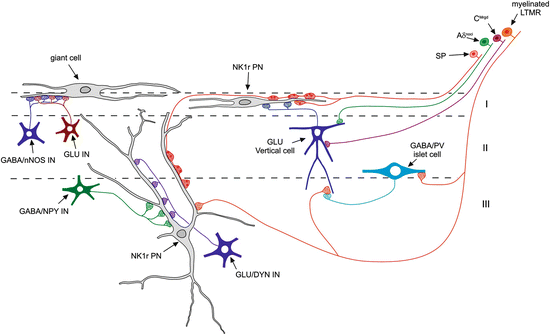
Fig. 2
A diagram illustrating some of the synaptic connections that have been identified in laminae I–III of the rodent dorsal horn. Three anterolateral tract projection neurons are indicated: a lamina I giant cell and projection neurons (PN) in laminae I and III that express the neurokinin 1 receptor (NK1r). Both lamina I and lamina III NK1r-expressing cells receive numerous synapses from peptidergic primary afferents that contain substance P (SP). The NK1r-expressing lamina I PN receives excitatory synapses from glutamatergic (GLU) vertical cells in lamina II, which are thought to be innervated by Aδ nociceptors (Aδnoci), non-peptidergic C fibre nociceptors (CMrgd) and myelinated low-threshold mechanoreceptors (LTMR). The myelinated LTMRs also innervate GABAergic islet cells that contain parvalbumin (PV), and they receive axoaxonic synapses from the PV cells. The lamina III PNs are selectively innervated by two distinct classes of interneuron: inhibitory cells that express neuropeptide Y (NPY) and excitatory (glutamatergic) cells that express dynorphin. The giant lamina I projection neurons appear to receive little or no direct primary afferent input but are densely innervated by excitatory and inhibitory interneurons. Many of the latter contain neuronal nitric oxide synthase (nNOS). For further details, see text
As stated above, the majority of ALT projection neurons in laminae I and III express the NK1r. These cells are densely innervated by substance P-containing primary afferent nociceptors, which provide around half of the glutamatergic synapses on their cell bodies and dendrites (Polgár et al. 2010; Baseer et al. 2012). This generates a powerful input and is likely to underlie the nociceptive responses of these cells. The remaining glutamatergic synapses on the projection neurons are presumably derived mainly from excitatory interneurons, and these are thought to include the vertical cells in lamina II (Lu and Perl 2005; Cordero-Erausquin et al. 2009). One of the functions of the excitatory interneurons that innervate lamina I projection neuron is to provide polysynaptic input from Aβ low-threshold mechanoreceptors (LTMRs). This contributes low-threshold components to the receptive fields of some of the projection cells (Bester et al. 2000; Andrew 2009), and loss of inhibition is thought to strengthen this indirect low-threshold pathway, leading to tactile allodynia in chronic pain states (Torsney and MacDermott 2006; Keller et al. 2007; Lu et al. 2013) (see below). We have recently found that lamina II vertical cells receive numerous contacts from myelinated LTMRs, which suggests that they may provide a disynaptic link between these afferents and lamina I ALT neurons (Yasaka et al 2014).
Unlike the NK1r-expressing projection neurons, the giant cells in lamina I apparently receive little or no primary afferent input, but these cells also respond to noxious stimuli, which are probably transmitted by polysynaptic pathways involving excitatory interneurons (Polgár et al. 2008).
There is evidence that different classes of ALT projection neuron are selectively innervated by specific populations of interneurons. For example, the large lamina III ALT neurons with prominent dorsal dendrites receive around one-third of their inhibitory synapses from NPY-containing GABAergic interneurons, whereas NPY is present in between 5 and 15 % of all GABAergic axons in laminae I–III (Polgar et al. 2011) (Fig. 3). These inhibitory inputs are apparently derived from a small subset of NPY-containing inhibitory interneurons, as both the size of the boutons and the intensity of NPY-immunoreactivity are considerably higher than those in the general population of NPY axons. In addition, the lamina III ALT neurons are also targeted by excitatory interneurons that express the opioid peptide dynorphin (Baseer et al. 2012). The giant cells in lamina I are also selectively innervated by inhibitory interneurons but this time by GABAergic cells that contain nNOS (Puskár et al. 2001). At present, less is known about the inhibitory inputs to the NK1r+ lamina I ALT neurons, although we have found that in the mouse, some of these cells are also innervated by nNOS-containing GABAergic axons (N Baseer and AJ Todd, unpublished data).
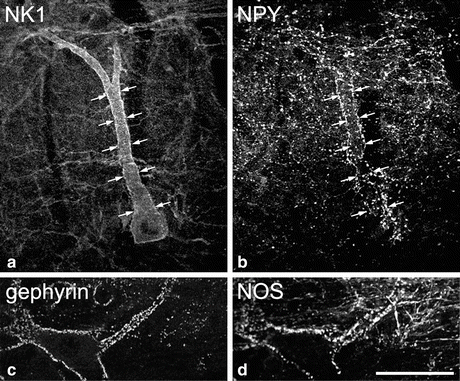
Fig. 3
Selective innervation of projection neurons by different types of inhibitory interneuron. Panels (a, b) show part of a parasagittal section of rat dorsal horn scanned to reveal the neurokinin 1 receptor (NK1) and neuropeptide Y (NPY). (a) The cell body and dorsal dendrite of a large NK1 receptor-immunoreactive lamina III projection neuron are visible. (b) NPY-containing axons, which are derived from local inhibitory interneurons, form a plexus in laminae I and II, and there is a dense cluster of these axons that contact the projection neuron. Arrows indicate corresponding locations in the two images. Panels (c, d) show part of a horizontal section through lamina I stained for the inhibitory synapse-associated protein gephyrin and the neuronal form of nitric oxide synthase (NOS). Gephyrin puncta (corresponding to inhibitory synapses) are scattered throughout lamina I and outline giant projection cells, one of which is seen here. The neuron is surrounded by numerous NOS-containing axons, again derived from local inhibitory interneurons. Scale bar = 50 μm for all parts (Parts a and b modified from Polgár et al. (1999), with permission from the Society of Neuroscience; parts c and d modified from Puskár et al. (2001), with permission from Elsevier)
It is likely that presynaptic inhibitory circuits are also arranged in a highly selective way. Peptidergic primary afferents receive few axoaxonic synapses, whereas these are found in moderate numbers on non-peptidergic nociceptors and are frequently associated with myelinated LTMRs. The axons that form synapses with the non-peptidergic nociceptors are enriched with GABA but not glycine, while those associated with Aδ D-hair afferents contain high levels of both GABA and glycine, indicating that they originate from different populations of inhibitory interneurons (Todd 1996). It has recently been shown that the parvalbumin-containing interneurons selectively innervate myelinated LTMRs (Hughes et al. 2012), since three-quarters of parvalbumin-immunoreactive axons in the inner part of lamina II (IIi) formed axoaxonic synapses on the central terminals of type II synaptic glomeruli (Ribeiro-da-Silva and Coimbra 1982), which are thought to be derived from Aδ D-hair afferents. It is not yet known which cells are responsible for presynaptic inhibition of the non-peptidergic C nociceptors.
1.6 Normal Function of Inhibitory Mechanisms
Inhibitory interneurons in the dorsal horn are thought to perform several different functions. For example, Sandkuhler (2009) has identified four specific mechanisms that are involved in the control of pain: (1) attenuation of the nociceptive inputs to dorsal horn neurons to achieve an appropriate level of activation in response to painful stimuli; (2) muting, to prevent spontaneous activity in neurons (including projection cells) that are driven by nociceptors; (3) separating different modalities, in order to prevent crosstalk that might lead to allodynia; and (4) limiting the spatial spread of sensory inputs, in order to restrict sensation to somatotopically appropriate body regions. Failure of each of these mechanisms would be expected to lead to the various symptoms that are seen in chronic pain states: hyperalgesia, spontaneous pain, allodynia and radiating/referred pain, respectively. Additional roles include the inhibition of itch, in response to counter-stimuli such as scratching (Davidson et al. 2009; Ross et al. 2010; Akiyama et al. 2011), and the sharpening of tactile acuity, by surround inhibition of LTMR afferents.
Since there are apparently many different populations of inhibitory interneurons in the dorsal horn, it is not likely that each of these functions is performed by a single population. However, different interneuron populations probably have a specific range of functions. For example, since many nNOS- and NPY-containing GABAergic neurons respond to noxious stimuli (Polgar et al. 2013b), those that innervate projection neurons in laminae I and III are likely to have a role in attenuating nociceptive inputs. In contrast, the parvalbumin interneurons, which generate presynaptic inhibition of myelinated LTMR inputs, may be involved in maintaining tactile acuity and preventing tactile allodynia (Hughes et al. 2012). Mice lacking the transcription factor Bhlhb5 show exaggerated itch that is associated with loss of inhibitory interneurons from the dorsal horn (Ross et al. 2010). We have recently found that there is a highly selective loss of nNOS- and galanin-containing inhibitory interneurons from these mice, which suggests that one or both of these populations is responsible for scratch-mediated inhibition of itch (Kardon et al 2014).
2 Plasticity of Inhibition in Neuropathic Pain States
Peripheral nerve injuries frequently give rise to neuropathic pain, which is characterised by spontaneous pain, allodynia and hyperalgesia. There are two lines of evidence to suggest that loss of inhibition in the spinal dorsal horn contributes to neuropathic pain. Firstly, suppressing inhibition by intrathecal administration of antagonists acting at GABAA or glycine receptors produces signs of tactile allodynia and hyperalgesia, which resemble those seen after nerve injury (Yaksh 1989; Sivilotti and Woolf 1994; Miraucourt et al. 2007; Lu et al. 2013). Secondly, a few studies have provided direct evidence for a loss of inhibitory synaptic transmission, by showing reduced inhibitory postsynaptic currents or potentials (IPSCs, IPSPs) after nerve injury (Moore et al. 2002; Scholz et al. 2005; Yowtak et al. 2011; Lu et al. 2013) (see below).
2.1 Animal Models of Neuropathic Pain
Before discussing the evidence for plasticity of inhibition in neuropathic pain, it is first necessary to give a brief account of the animal models that have been used to investigate this issue. Complete transection of a peripheral nerve in humans generally leads to an area of anaesthesia, whereas partial nerve injuries are more likely to give rise to symptoms and signs of neuropathic pain. Based on this observation, several partial nerve injury models have been developed in rodents, and some of the most commonly used ones will be mentioned here. Bennett and Xie (1988) reported that loose ligation of the sciatic nerve led to oedema and subsequent self-strangulation of the nerve, similar to an entrapment neuropathy. Animals that have undergone this procedure, which is known as chronic constriction injury (CCI), gradually develop thermal and mechanical hyperalgesia, together with tactile allodynia. These are maximal at ~2 weeks after surgery and last for around 2 months (Attal et al. 1990). A disadvantage of the method is the potential variability that results from unavoidable differences in the tightness of the ligatures, and application of a polyethylene cuff around the nerve has therefore been used as an alternative. Kim and Chung (1992) developed the spinal nerve ligation (SNL) model, which involves tight ligation of the L5 (and optionally the L6) spinal nerve(s). This leads to a rapid and long-lasting tactile allodynia and thermal hyperalgesia. Another technique, spared nerve injury (SNI), involves tight ligation of two of the three major branches of the sciatic nerve (tibial and common peroneal), leaving the remaining branch (sural) intact, and results in a rapid onset of tactile allodynia, with cold allodynia and a moderate degree of heat hyperalgesia (Decosterd and Woolf 2000).
2.2 Reduced Inhibitory Synaptic Transmission in Neuropathic Pain States
Electrical stimulation of dorsal roots in spinal cord slice preparations normally evokes (polysynaptic) IPSCs in dorsal horn neurons, due to activation of inhibitory interneurons that are presynaptic to the recorded cell. Moore et al. (2002) reported that the proportion of lamina II neurons showing these evoked IPSCs (eIPSCs) was reduced in CCI and SNI models but was unchanged in slices from rats that had undergone complete transection of the sciatic nerve (when compared to slices from unoperated animals). In addition, the amplitude and duration of eIPSCs was reduced in both CCI and SNI models. There was also a reduction in the frequency of miniature IPSCs (mIPSCs; i.e. those recorded in the presence of tetrodotoxin to block synaptic activity) in both neuropathic pain models. Similar results were reported by the same group in a subsequent study (Scholz et al. 2005), while Yowtak et al. (2011) found a reduction in mIPSC frequency (but not amplitude) in the SNL model.
The reduced frequency of mIPSCs indicates a presynaptic mechanism involving the inhibitory interneurons, and this is consistent with immunocytochemical evidence that there is no reduction of GABAA receptors in the superficial dorsal horn (Moore et al. 2002; Polgár and Todd 2008). Although it has been reported that there is a loss of GABAB receptors from the L5 dorsal root ganglion after SNL (Engle et al. 2012), this would have no effect on IPSCs in the dorsal horn.
2.3 Possible Mechanisms for Reduced Inhibition Following Peripheral Nerve Injury
Several different mechanisms have been proposed to explain loss of inhibition in the superficial dorsal horn after nerve injury. Some of these are consistent with the reduced mIPSC frequency, for example, death of inhibitory interneurons and reduced neurotransmitter release. Other mechanisms that have been suggested would not account for the change in mIPSCs, although these could still contribute to neuropathic pain. These will be discussed in the following sections.
2.3.1 Loss of Inhibitory Interneurons
One mechanism that could result in loss of inhibition would be death of inhibitory interneurons, and numerous studies have addressed this issue. There has been considerable controversy over whether or not there is significant neuronal death in the dorsal horn after peripheral nerve injury and, if so, whether this affects inhibitory interneurons. Three approaches have been used to investigate these questions: (1) detection of apoptotic cell death, (2) assessment of the number of neurons in each lamina after nerve injury and (3) analysis of the numbers of GABAergic neurons, by using immunocytochemistry, in situ hybridisation for glutamate decarboxylase (GAD, the enzyme that synthesises GABA) or mouse lines in which these GABAergic neurons express GFP. It should be noted that there are two isoforms of GAD, named from their molecular weights: GAD65 and GAD67, and both of these are present in the dorsal horn (Mackie et al. 2003).
Several studies have reported apoptosis in the dorsal horn following peripheral nerve injury, based on staining with the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labelling (TUNEL) method (Kawamura et al. 1997; Azkue et al. 1998; Whiteside and Munglani 2001; Moore et al. 2002; Polgár et al. 2005; Scholz et al. 2005) or detection of the activated form of caspase-3 (Scholz et al. 2005), which is thought to lead to inevitable apoptotic cell death. In some of these studies, no attempt was made to identify whether the apoptotic nuclei belonged to neurons, but Azkue et al. (1998) concluded that TUNEL-positive cells were neuronal, based on expression of a cytoskeletal marker, while Moore et al. (2002) reported that 10 % of them were immunoreactive for the neuronal marker NeuN. In addition, Scholz et al. (2005) observed extensive co-localisation of activated caspase-3 and NeuN after SNI. Polgár et al. (2005) also looked for evidence of neuronal apoptosis in the SNI model but found that although there were numerous TUNEL-positive cells in the ipsilateral dorsal horn, these were virtually all associated with the calcium-binding protein Iba-1, a marker for microglia. They also found no coexistence between NeuN and either TUNEL staining or activated caspase-3 in the SNI model, despite the fact that NeuN co-localised with both these markers in the developing olfactory bulb, where neuronal apoptosis is known to occur. This indicates that failure to detect coexistence in the spinal cord was unlikely to result from early loss of NeuN, making neurons undetectable. Polgár et al. (2005) also observed that while TUNEL-positive nuclei were present in the ipsilateral dorsal horn, they were also found in relatively large numbers in the adjacent white matter, including the dorsal columns, where neurons are seldom present.
There has also been disagreement as to whether the numbers of neurons in the dorsal horn are altered after nerve injury. Scholz et al. (2005) observed a ~20 % loss of neurons from laminae I to III 4 weeks after SNI, whereas two other studies observed no loss of neurons from this region in either the SNI or CCI models (Polgár et al. 2004, 2005). All of these studies used stereological methods to assess the packing density of NeuN-immunoreactive profiles, and it is therefore difficult to explain the discrepancy.
There has also been considerable controversy over whether the numbers of GABAergic neurons are reduced after peripheral nerve injury. Two early studies reported a dramatic (~80–100 %) loss of GABA-immunostaining after CCI (Ibuki et al. 1997; Eaton et al. 1998). Surprisingly, both of these studies also observed a substantial loss of GABA on the contralateral side, even though signs of neuropathic pain are generally not present on this side. Scholz et al. (2005) reported a 25 % reduction in the number of cells with GAD67 mRNA after SNI, although this is not consistent with the earlier report from this group that GAD67 protein and mRNA levels were not altered in this model (Moore et al. 2002). In complete contrast, Polgár et al. (2003) found no alteration in immunostaining for either GABA (Fig. 1) or glycine in the ipsilateral or contralateral dorsal horn of animals that had undergone CCI and which showed clear signs of thermal hyperalgesia. Specifically, the proportions of neurons that were GABA- and/or glycine-immunoreactive on either side of CCI rats did not differ from those in naïve animals. Again, this discrepancy is difficult to interpret. However, immunocytochemistry for the amino acids is technically challenging, requiring rapid and efficient fixation with a relatively high concentration of glutaraldehyde. Technical issues associated with retention of GABA are likely to underlie the differences between the immunocytochemical studies.
A recent study by Yowtak et al. (2013) examined the effects of SNL on the GIN mouse (see above) and reported a reduction of ~30 % in the number of GFP+ cells in the lateral part of lamina II in the L5 segment. Since around a third of GABAergic cells are GFP+ in this line (Heinke et al. 2004), they interpreted this result as indicating a loss of GABAergic neurons. However, it is possible that this reflects downregulation of GFP, rather than cell loss. In addition, the significance of this finding is difficult to interpret, since the signs of neuropathic pain in the SNL model depend on inputs from the intact L4 spinal nerve (Todd 2012), which does not innervate the lateral part of the L5 segment (Shehab et al. 2008).
Taken together, these studies indicate that there is a great deal of controversy over whether there is any neuronal loss after peripheral nerve injury and, if so, whether this involves inhibitory interneurons. Importantly, in the studies that found no neuronal apoptosis or loss of GABAergic neurons, there were clear signs of neuropathic pain (Polgár et al. 2003, 2004, 2005), similar to those found in the original reports of these models. This strongly suggests that loss of GABAergic neurons is not required for the development of neuropathic pain after nerve injury.
Related posts:





Full access? Get Clinical Tree


