183 Nonsteroidal Antiinflammatory Agents
Nonsteroidal antiinflammatory drugs (NSAIDs) have important clinical uses in selected critically ill patients for treatment of pain and inflammatory states or reduction of fever.1–3 However, drugs in this class can cause serious side effects and/or affect other medications used concomitantly. The pharmacologic characteristics of NSAIDs related to cyclooxygenase-1 (COX-1) and COX-2 enzyme inhibition may result in severe gastrointestinal, cardiovascular, and renal side effects.4–8 Two highly COX-2-selective agents have been withdrawn from the market as a result of toxicity.9 Patient characteristics and differences in NSAID toxicity profiles are important for the judicious use of NSAIDs in the critically ill patient.
 NSAID Pharmacodynamics
NSAID Pharmacodynamics
NSAIDs include aspirin, indomethacin, ibuprofen, naproxen, diclofenac, and a product which is relatively more selective for the COX-2 isoform of cyclooxygenase (i.e., celecoxib). All NSAIDs have analgesic, antiinflammatory, and antipyretic properties. NSAIDs belong to a number of chemical families including acetic acids, oxicams, propionic acids, salicylates, fenamates, furanones and coxibs (Table 183-1). All NSAIDs are weakly acidic chemical compounds and share similarities in pharmacokinetic properties.10 Their absorption is primarily in the large surface area of the small intestine as well as in the stomach.10 Gastrointestinal absorption of NSAIDs occurs rapidly, usually within 15 to 30 minutes. Different product formulations, including enteric-coated and delayed-release preparations, decrease gastric emptying, and altered gastric transit time can delay drug absorption and time to peak effect.11 For example, 4 to 6 hours is required for peak absorption of enteric-coated products.11 After absorption, NSAIDs are more than 90% bound to albumin, which influences their distribution and drug-drug interaction potential. Hypoalbuminemia (e.g., due to alcoholic liver disease) can result in greater unbound drug and increased risk for NSAID-related adverse events.11
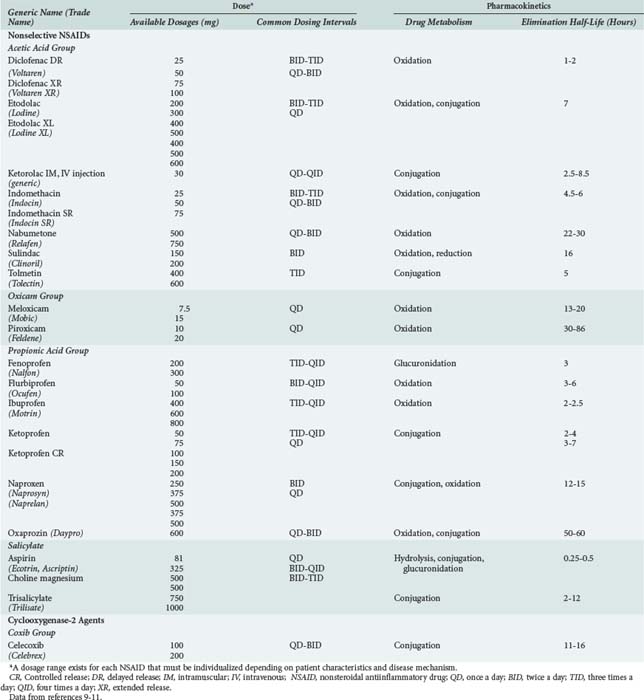
NSAIDs are primarily eliminated by renal and biliary excretion.12 The elimination half-lives of NSAIDs vary from 0.25 to 86 hours, which accounts for differences in dosing schedules (see Table 183-1).11 Factors that delay NSAID clearance increase their potential for adverse reactions. Reduced renal function prolongs NSAID half-life, and the dose should be lowered proportionally in patients with impaired kidney function.10,12 Some NSAIDs are hepatically metabolized to both active and inactive metabolites, primarily through the cytochrome P450 enzymes, glucuronidase enzymes, or both.10,13 Nabumetone and sulindac are prodrugs and require metabolism by the liver to generate pharmacologically active metabolites.10 Moderate to severe liver disease impairs NSAID metabolism, increasing the potential for NSAID toxicity. With advanced age, the hepatic clearance of diclofenac, etodolac, flurbiprofen, ibuprofen, indomethacin, meloxicam, nabumetone, naproxen, oxaprozin, piroxicam, and sulindac is slower because of decreased hepatic phase I oxidative, reductive, and hydrolytic catalytic reactions.10
NSAIDs have a number of physiologic effects, although their principal action is inhibition of the cyclooxygenase enzyme.1,2 COX is responsible for the production of prostaglandins (PG) and thromboxanes (TX), which are derived from arachidonic acid, an unsaturated fatty acid present in all body cell membranes. PGs and TXs mediate normal homeostatic functions of the upper gastrointestinal tract, kidneys, endothelium, vascular smooth muscle, and platelets, among other tissues and organs. PG and TX are critical in the inflammatory response because of their influences on vascular permeability, platelet function, and immune reactions. These autocoids are involved in both peripheral and central pain processing and have a role in fever production.14,15
As noted earlier, there are two isoforms of the COX enzyme: COX-1 and COX-2. Although the COX enzymes are coded on two separate genes, they share 63% structural homogeneity, have similar mechanisms of action, and produce identical compounds from arachidonic acid.14–17 Expression and regulation of COX-1 and COX-2 differ in various organs and tissues; however, their physiologic effects are overlapping.2,14,15,18 Differences in the structural configurations of COX-1 and COX-2 enzyme side chains determine whether a particular NSAID will inhibit the enzyme. Isoform nonselective NSAIDs (e.g., naproxen, ibuprofen) inhibit both COX-1 and COX-2, thereby decreasing production of PGs involved in both homeostatic and inflammatory actions. The COX-2-selective agent, celecoxib, is 50-fold more active against COX-2 than COX-1 and therefore exerts actions primarily in inflammatory processes.18,19 Both COX-1 and COX-2 are involved in pain processing.
Differences in COX-1 and COX-2 inhibition allow for comparisons of drug effect.6 Nonselective NSAIDs exert one of three kinetic models for inhibiting COX-1 and COX-2: (1) rapid, reversible binding (e.g., ibuprofen); (2) rapid, lower-affinity reversible binding followed by time-dependent, higher-affinity, slowly reversible binding (e.g., indomethacin); or (3) rapid, irreversible binding followed by covalent modification (e.g., aspirin). Aspirin is the only NSAID that covalently modifies both COX enzymes, thereby resulting in permanent inhibition of both isoforms.20 COX-2-selective inhibitors act on COX-2 by a time-dependent, slowly reversible mechanism. They also can affect COX-1 by a freely reversible and competitive mechanism. The result of this two-stage process by COX-2-selective agents is maximal inhibition of COX-2 with minimal inhibition of COX-1.16,17,21
The biochemical selectivity of NSAIDs is related to the in vitro drug concentration necessary to inhibit COX-2 activity completely and COX-1 activity by 50%.2,15 Data from clinical trials demonstrating a decreased incidence of gastrointestinal toxicity and an absence of platelet inhibition with COX-2-selective versus nonselective agents have been the clinical parameters used to distinguish among the various agents.2
COX converts arachidonic acid to the inactive precursor, PGG2, and then PGH2. PGH2 is metabolized in various tissues to physiologically active products including PGI2, PGE2, and TXA2. The concentration of different PGs determine their biological effects on tissues. PGs exert their effects by activating cell-membrane receptors of the superfamily of G protein–coupled receptors.1 PGI2 has important regulatory effects on renal blood flow, gastric mucosa, uterine smooth muscle, and bronchial smooth muscle. PGI2 also inhibits platelet aggregation. PGE2 is an abundant PG with important regulatory effects on fever and on the reproductive, gastrointestinal, neuroendocrine, and immune systems.22 PGE2 is present at sites of inflammation as a potent vasodilator in acute and chronic inflammatory diseases and in tissue injury. PGE2 also can promote labor and dysmenorrhea.22 TXA2 promotes platelet aggregation and vasoconstriction. TXA2 is released with tissue injury and plays a role in cellular responses to inflammation.22
PGs produced by COX-1 are primarily involved in maintaining the protective gastrointestinal mucosal barrier in the stomach and intestines, modulating intrarenal hemodynamics, influencing platelet function (especially aggregation), and regulating vascular homeostasis. For example, PGs produced by COX-1 provide gastric protection by reducing gastric acid secretion, stimulating mucus secretion, and promoting gastric mucosa vasodilation. Kidney function is affected by the localization of COX-1 in the collecting ducts and renal vasculature. COX-1 converts PGH2 to TXA2, which promotes platelet aggregation. Although the major role of COX-1 is homeostasis, COX-1 may contribute to PG production in certain inflammatory reactions, including those in the synovia of inflamed joints and atherosclerotic plaques.14,15,18
The primary role of COX-2 is in inflammatory reactions that result in PG production by fibroblasts, macrophages, endothelial cells, and synoviocytes. This enzyme is also important in pain and fever mechanisms.16 Certain other tissues express COX-2, especially the cortical macula densa, medullary interstitial cells, and the kidney vasculature.15 Small amounts of COX-2 are also found in the small intestine, ovary, uterus, bone, and brain.14,16 Because COX-2 expression is regulated by growth factors, its role in wound repair is under investigation.17
 Clinical Implications and Uses of NSAIDs in Critically Ill Patients
Clinical Implications and Uses of NSAIDs in Critically Ill Patients
Although NSAIDs have important analgesic, antiinflammatory, and antipyretic activity, their role in critically ill patients should be limited, owing to their potential for toxic side effects.23 In clinical trials, all nonselective and COX-2-selective NSAIDs in equipotent doses have demonstrated similar efficacy in relieving pain, inflammation, and fever.24–26 However, there is significant variability in the clinical effects of NSAIDs within and among patients, with approximately 70% to 80% of individuals responding to any particular agent.25 Lack of response to one NSAID does not preclude benefit from another.25 Differences in NSAIDs may be related to COX-1 and COX-2 inhibitory pharmacodynamics, because no definite clinical characteristics have been identified in nonresponders compared to responders.10,20 In contrast, toxicity profiles of nonselective and COX-2-selective agents have a defined relationship to the degree of COX-1 and COX-2 inhibition, especially for the development of gastrointestinal adverse events or antiplatelet effects.
Management of Pain and Inflammation
Pain management is a key issue for critical care clinicians. Although morphine is the primary analgesic of choice among critically ill patients with pain, NSAIDS may have a role in selected patients.26 Pain is initiated by activation of tissue nociceptors in various disease states by mechanical, thermal, and chemical stimuli. Surgical trauma and other forms of tissue injury induce expression of COX-2, and to a lesser extent COX-1, resulting in generation of PGs, especially PGE2.27–29 PGs sensitize A-δ and C primary afferent sensory nerve fibers that carry impulses to the dorsal horn of the spinal cord. Glutamate, substance P, and other mediators along with PGs are involved in dorsal horn pain processing.30 By inhibiting PGs at these different levels of the pain matrix, NSAIDs can have important effects on pain processing. For example, NSAIDs reduce PG-mediated protein kinase A phosphorylation of sodium channels in nociceptor terminals.
Surgical Pain
Inadequate postoperative pain control has been associated with increased morbidity, increased length of stay, and increased costs for intensive care unit (ICU) patients.31,32 Opioids are commonly prescribed for surgical and trauma pain and other acute pain states, but adverse events such as nausea and vomiting, drowsiness, and respiratory depression may limit their use in some patients and prolong postoperative recovery and increase costs.31 NSAIDs are effective in combination with other analgesics in managing the acute pain of tissue injury.33 For example, the use of NSAIDs in orthopedic and other types of surgeries including knee arthroscopy, hip replacement, spinal surgery, and gynecologic laparoscopy decreases postoperative opioid requirements.24,26,34–38 In addition, NSAID therapy alone can provide effective postoperative pain relief.31,38,39 However, because of the potential for adverse reactions (e.g., gastrointestinal toxicity, platelet inhibition with increased bleeding risk, renal dysfunction), especially with hypovolemia, NSAIDs should be used cautiously in the management of postoperative pain.24,31,39 Although COX-2-selective inhibitors are equally effective in pain relief as nonselective NSAIDs, they should be used cautiously in the ICU owing to their potential for cardiac and gastrointestinal toxicity.39 Recent studies of the use of COX-2-selective inhibitors in orthopedic and gynecologic surgeries demonstrated significant reductions in opioid consumption, decreased postoperative opioid side effects including nausea and vomiting, and improved subjectively.24,28,40,41 In addition, preoperative use of COX-2-selective agents in knee arthroscopy, compared with postoperative use, can delay the time to first analgesic request and decrease total opioid consumption.35 The clinical impact of NSAID therapy on wound and bone healing after surgery is unclear.42,43 NSAIDs, especially indomethacin, have been used perioperatively to reduce heterotopic bone formation after acetabular fracture surgery.44 Recent data from animal and human trials suggest that NSAIDs may impair bone healing after fractures because of the role of PGs in osteogenesis.42,43,45
Ketorolac is currently the only injectable NSAID available in the United States. Clinical studies comparing injectable ketorolac with morphine in managing postoperative pain after orthopedic, gynecologic, and major abdominal procedures have shown similar efficacy but slower onset of action with ketorolac.46 Combination therapy with ketorolac and morphine may provide analgesic benefit compared with morphine alone and reduce total morphine consumption.47 However, use of ketorolac in acute pain states is now limited because of postmarketing reports of toxicity including peptic ulcers, gastrointestinal bleeding, and renal insufficiency.46 In addition, dosing adjustments are necessary for patients with renal dysfunction and elderly patients, and duration of therapy never should exceed 5 days.48
Regional Inflammatory States
The antiinflammatory properties of NSAIDs are beneficial in specific acute disease states affecting critically ill patients, including systemic and regional rheumatic disorders and localized inflammatory conditions such as pleuropericarditis. PGs, especially PGI2 and PGE2, are induced by interleukin (IL)-1, IL-6, and IL-8. These PGs are important mediators of inflammatory reactions; they influence vascular reactivity and increase vascular permeability.22
Rheumatic diseases such as rheumatoid arthritis and systemic lupus erythematosus (SLE) present therapeutic challenges to the critical care clinician. Concerns about the role of NSAIDs in causation of certain acute problems, especially gastrointestinal bleeding and fluid retention, have arisen. NSAIDs are an important component of the therapeutic regimen for the synovitis of inflammatory arthritis and for serositis involving pleural or pericardial membranes.48,49 Discontinuation of NSAID therapy can result in a significant increase in synovitis. The antiinflammatory effects of NSAIDs often require higher doses than those needed for a chronic analgesic response. No significant differences in effectiveness in suppressing inflammation have been demonstrated among the various nonselective and COX-2-selective agents.24,25,50 Acute crystal-induced arthritis such as gout and pseudogout, although uncommon, still occasionally present in critically ill patients. Inflammation in response to uric acid and calcium pyrophosphate dihydrate crystals, respectively, is induced by immune mediators such as PGs, cytokines, bradykinin, and leukotrienes, which produce capillary dilation, neutrophil migration, and pain stimulation. NSAIDs, especially indomethacin, have been shown to be beneficial in acute crystal-induced arthritis.51 Aspirin should not be used for gout because it influences renal tubular uric acid excretion, thereby causing fluctuations in serum uric acid levels, potentially aggravating acute gouty arthritis. Because all NSAIDs except ketorolac are administered only orally and can be toxic, their use is limited in the critical care setting. Alternative treatments for crystal-induced arthritis include corticosteroids and colchicine for gout.
Occasionally, acutely ill patients require NSAID therapy for their antiinflammatory and analgesic properties because of concurrent regional musculoskeletal disorders such as shoulder or elbow tendonitis or back problems.52 Finally, NSAIDs have been used to treat pleuropericarditis of nonrheumatic origin, including viral serositis and postmyocardial infarction syndrome.
Cardiovascular Protection
Abnormalities within the cardiovascular system are frequently present in critically ill patients. Because PGs play pathophysiologic roles in coagulation and inflammatory mechanisms involved in cardiovascular diseases, NSAIDs, especially aspirin, have an important therapeutic role. TXA2, synthesized by platelets, is produced by COX-1 (together with another enzyme, thromboxane synthase); it promotes platelet aggregation on abnormal vascular endothelium and in areas of vascular stasis, leading to thrombosis. All nonselective NSAIDs inhibit COX-1; however, only aspirin does so irreversibly through acetylation.53 The U.S. Preventive Services Taskforce Report strongly recommended the use of aspirin in adult patients who are at risk for coronary heart disease.54 Practice guidelines recommend the use of low-dose aspirin (81-325 mg/d) for high-risk patients (i.e., 5% risk within 5 years) to reduce cardiovascular events including nonfatal myocardial infarction, fatal coronary heart disease, and nonhemorrhagic stroke.53,55 For those individuals with a lower risk of having a coronary heart disease event, the benefit was negated by the toxic effects of aspirin, including gastrointestinal events and hemorrhagic stroke. An update of the evidence from the U.S. Preventive Task Force was published in 2009.55 Following a systematic review of randomized clinical trials, the authors concluded that aspirin reduces the risk of coronary vascular disease (CVD) in adults without a history of CVD.55 Critical care clinicians should consider using aspirin for patients at risk for CVD; however, the risks must be weighed against the benefits because of the increased risk for gastrointestinal bleeding events.55
Although other nonselective NSAIDs reversibly inhibit the COX-1 enzyme in platelets, they have not been demonstrated to reduce cardiac events and currently should not be used for primary or secondary prevention of vascular disease. COX-2-selective agents do not have demonstrated cardiovascular benefits.56
Aspirin has been compared with adjusted-dose warfarin for the prevention of stroke in atrial fibrillation. The 8th American College of Chest Physicians (ACCP) Consensus Conference on Antithrombotic Therapy recommended that drug selection in atrial fibrillation be based on risk likelihood: adjusted-dose warfarin for patients with a high risk of stroke, aspirin or adjusted-dose warfarin for those with a moderate risk; and aspirin for those with a low risk.53 The risk for stroke is increased by a history of prior stroke, systemic embolus, hypertension, poor left ventricular systolic function, age older than 75 years, rheumatic mitral valvular disease, or a prosthetic heart valve.53 Although both adjusted-dose warfarin and aspirin confer significant stroke reduction in the case of atrial fibrillation, their concomitant use does not impart greater benefit, and increased side effects do occur.53
Aspirin therapy, alone or in combination with dipyridamole, can delay progression of established arterial occlusive disease in patients with chronic lower-extremity arterial insufficiency.53 Low-dose aspirin is beneficial in preventing morbidity and mortality from stroke and myocardial infarction in patients with peripheral arterial disease.53,57,58 In addition, low-dose aspirin is used as prophylaxis and treatment for ischemic cerebrovascular disease, alone and in combination with dipyridamole, and has been demonstrated to reduce the risk of stroke in individuals with transient ischemic attacks or completed ischemic strokes due to thrombosis.53
Fever
The mechanisms of fever involve either peripheral release of pyrogenic cytokines (IL-1, IL-6, tumor necrosis factor [TNF], and interferon [IFN]-α) from monocytes and macrophages or the presence of circulating endotoxins which stimulate central production of PGE2 via COX-2 in vascular endothelial cells. PGE2 targets hypothalamic thermoregulatory neurons.59–61 Knockout mice lacking the COX-2 enzyme are unable to mount a fever when exposed to exogenous pyrogens.59,60 The COX-1 enzyme is not involved in thermal regulatory control.59
Aspirin has long been recognized as an effective antipyretic agent, and it is the gold standard with which acetaminophen and other NSAIDs are compared. Studies evaluating the antipyretic properties of NSAIDs are primarily from the pediatric literature. A meta-analysis of adult fever trials has not been possible because of differences in patient populations, NSAID dosing schedules, and outcome measures. However, studies in adults have shown equal or superior antipyretic efficacy of NSAIDs compared with acetaminophen.60 NSAIDs are more effective in reducing fevers associated with cancer than those caused by infection, although the mechanism is unclear.60 Duration of action of various NSAIDs in fever is related to drug half-life and drug concentration in the hypothalamus, a parameter determined by drug transport across the blood-brain barrier.59 There are limited data evaluating clinical use of COX-2-selective agents for fever, but a beneficial effect has been demonstrated.62,63,64 Toxicities of the various nonselective NSAIDs limit their clinical utility compared with acetaminophen, especially in critically ill patients.60,65
Septic Shock
Ibuprofen and other NSAIDs have some physiologic effects in sepsis, although they do not reduce morbidity and mortality in septic shock, except possibly in patients with hypothermia.66,67 A prospective randomized trial evaluated the impact of ibuprofen on organ failure and mortality in 455 patients with sepsis syndrome.67 Significant improvements in temperature, heart rate, oxygen consumption, and lactic acidosis were noted, but there was no reduction in organ failure or 30-day mortality compared with standard care. The Surviving Sepsis Campaign guidelines do not list NSAIDs as a treatment option for septic shock.68



Full access? Get Clinical Tree


