Nitric Oxide Involvement in Migraines
Helle U. Iversen
Uwe Reuter
Over the years, many provoking factors have been studied in the search for migraine mechanisms (76). Glyceryl trinitrate (GTN) and histamine have been the most widely used compounds to induce and study headache; migraine patients experienced intense migraine-like headache whereas healthy subjects suffered mild headache (12,14,32,46,77,88). Both GTN and histamine were able to induce cluster attacks in cluster headache patients, but only during cluster periods (19,32). In healthy volunteers, GTN-induced headache is caused by nitric oxide (NO) or activation of the NO-cyclic guanosine monophosphate (cGMP) cascade (39). First, the long-acting nitrate, 5-isosorbide-mononitrate gives rise to NO formation and induces headache-like GTN, but its other metabolites are different from those of GTN (36). Second, N-acetylcysteine, which enhances the cardiac vasodilatory effects of GTN presumably by stimulating NO production, also potentiated GTN-induced headache (34). In more recent studies, both GTN and histamine induced a migraine headache about 5 hours after drug administration (51,94), suggesting a common trigger mechanism. GTN may induce mast cell degranulation (84), although not all agree (61). GTN-induced headache could be caused by histamine release, but mepyramine, an H1 antagonist that blocks histamine-induced headache (46,53), had no effect on intensity and characteristics of GTN-induced headache, migraine, or arterial responses (37,52) (Fig. 30-1). Histamine dilates large human cephalic arteries in vitro via stimulation of H1 receptors and release of NO (40,99). The action of NO, or activation of the NO-cGMP cascade, thus is likely to be the common mediator of headache and migraine induced by both GTN and histamine.
NO is an ubiquitous gas, and NO synthesizing enzymes, endothelial NO synthase (eNOS), neuronal NOS (nNOS), and inducible NOS (iNOS), have been found in most tissues including brain, meninges, trigeminal ganglion, endothelium, peripheral nerves, vascular smooth muscle, myocardium, macrophages, neutrophils, and glial cells (44,61). The production and release of NO can be stimulated via activation of 5-HT2B/C receptors, bradykinin, glutamate, acetylcholine, histamine, substance P, NMDA, and probably calcitonin gene-related peptide (CGRP) (9,23,26,27,29,57). In reverse, NO may be able to release neuropeptides such as CGRP or vasoactive intestinal peptide and thereby could play a role in neurogenic inflammatory reactions within meninges. For example, in pial vessels NO-induced vasodilation was blocked by a CGRP receptor antagonist (CGRP8-37) indicating a link between both molecules (103). Also, mechanical influence of sheer stress on the artery wall induces NO production (85).
NO production causes activation of soluble guanylate cyclase and subsequent increase in cGMP. This induces gene expression via a host of different transcriptional pathways such as MAP kinases or NFκB. Further detailed mechanisms of NO are discussed in Chapter 15. NO has been identified as a key molecule in several physiologic processes, including pain and sensitization. For example, NO-synthesizing enzymes are strongly upregulated in neurons after axotomy and thereby maintain hyperalgesia (106). In addition, NOS upregulation in the rostral ventral medulla has been reported in visceral pain models (102). Taken together, there is strong evidence for a pivotal role of NO in the genesis of migraine pain.
EFFECTS OF NITRIC OXIDE IN MIGRAINE PATIENTS
Migraine patients are more sensitive to the immediate effects of GTN, in terms of headache and arterial dilatation, than healthy subjects and tension-type headache patients (97; see Chapter 23). Upon exposing migraine patients with and without aura to GTN, an immediate headache of mild to moderate intensity appeared during infusion. After a delay of around 1 hour, the patients experienced their usual premonitory symptoms; a characteristic migraine attack occurred 5 to 6 hours after GTN challenge (1,12,96,98). In only one patient with migraine with aura was an aura
triggered (1). Migraine attacks were induced in 75% of the migraine patients and were remarkably reproducible (1). A similar response was observed after sublingual administration of the long-acting nitrate isosorbide dinitrate (4). The surprisingly long latency period between GTN/histamine exposure and a fully developed migraine attack indicates that activation of NO—or steps in the NO-cGMP cascade—initiate a somewhat slow process that results in an attack. GTN-induced migraine can be attenuated by steroids, which inhibit iNOS, the enzyme responsible for slow inducible NO production (16), and partly prevented by 2 weeks of valproate treatment before GTN exposure (Fig. 30-2) (101). NO acts by increasing GMP. The cGMP-hydrolyzing phosphodiesterase 5 (PDE5) inhibitor sildenafil, which acts further along the NO-cGMP cascade exclusively by increasing cGMP, induced migraine in 10 of 12 patients with migraine without aura. No effects on the middle cerebral artery were detected (47). This study showed that NO itself is not necessary to induce migraine, but a cGMP-dependent mechanism without initial dilatation of the middle cerebral artery appear to be implicated. Others have shown that sildenafil increased endothelium-dependent flow-mediated vasodilatation (42). These results raise several questions. Is the headache caused by a direct effect on perivascular sensory nerve terminals? Is it a central effect? Why was dilatation based on the trigeminoparasympathetic reflex not observed?
triggered (1). Migraine attacks were induced in 75% of the migraine patients and were remarkably reproducible (1). A similar response was observed after sublingual administration of the long-acting nitrate isosorbide dinitrate (4). The surprisingly long latency period between GTN/histamine exposure and a fully developed migraine attack indicates that activation of NO—or steps in the NO-cGMP cascade—initiate a somewhat slow process that results in an attack. GTN-induced migraine can be attenuated by steroids, which inhibit iNOS, the enzyme responsible for slow inducible NO production (16), and partly prevented by 2 weeks of valproate treatment before GTN exposure (Fig. 30-2) (101). NO acts by increasing GMP. The cGMP-hydrolyzing phosphodiesterase 5 (PDE5) inhibitor sildenafil, which acts further along the NO-cGMP cascade exclusively by increasing cGMP, induced migraine in 10 of 12 patients with migraine without aura. No effects on the middle cerebral artery were detected (47). This study showed that NO itself is not necessary to induce migraine, but a cGMP-dependent mechanism without initial dilatation of the middle cerebral artery appear to be implicated. Others have shown that sildenafil increased endothelium-dependent flow-mediated vasodilatation (42). These results raise several questions. Is the headache caused by a direct effect on perivascular sensory nerve terminals? Is it a central effect? Why was dilatation based on the trigeminoparasympathetic reflex not observed?
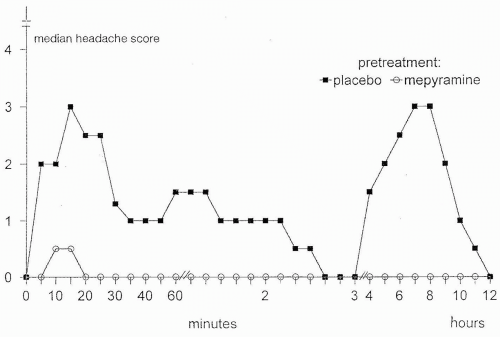 FIGURE 30-1. Histamine induced headache and migraine after pre-treatment with mepyramine 0.5 mg/kg or placebo. Mepyramine significantly blocked the histamine-induced migraine (51). |
Sicuteri et al. described first-degree relatives of migraine patients developing migraine after GTN, believing it only occurred in subjects with a genetic predisposition to migraine (91). Thus, migraine patients, and possibly first-degree relatives, are supersensitive to NO that initiates a process leading to a migraine attack (95). This appears
independent of the frequency of the patients’ naturally occurring migraine attacks (12).
independent of the frequency of the patients’ naturally occurring migraine attacks (12).
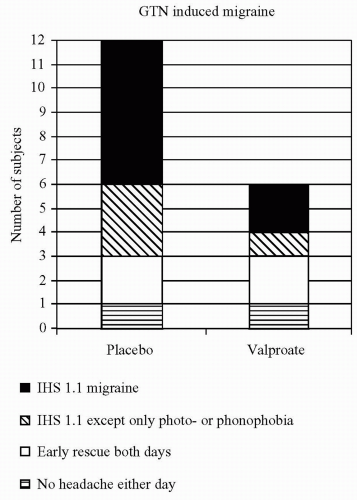 FIGURE 30-2. In 12 patients with migraine without aura, valproate 1000 mg, or placebo was given daily for 2 weeks. On the last treatment day, a 20-minute intravenous infusion of GTN 0.25 μg/kg per minute was given. 0 to 10. Valproate significantly decreased the GTN-induced headache (101). |
Migraine patients showed no altered systemic endothelial response compared to controls (96), and the frequency of migraine attacks did not depend on the basal plasma NO levels (67). A mild parasympathetic dysfunction may be involved and may be responsible for the observed denervation supersensitivity to NO in cerebral arteries (97). A greater increase in cGMP after intramuscular injection of a cholinergic agent occurred in peripheral venous blood (68). This may provide a link between parasympathetic hypofunction and arterial NO hypersensitivity in migraine, because cholinergic agents seem to liberate NO, at least from the endothelium (24). Direct activation of perivascular sensory nerve fibers (65) or central effects of NO may be other possibilities.
The similar frequency for migraine in boys and girls before adolescence, increased frequency in women thereafter, and the importance of the menstrual cycle, indicate a possible role of the sex hormones for expression of migraine attacks; sensitivity to NO is greater with higher levels of estrogen (73).
CGRP also may play an important role in the pathogenesis of migraine via activation of CGRP receptors in the trigeminovascular system. The CGRP-receptor antagonist, BIBN4096BS, has proven efficacy in the acute treatment of migraine attacks and represents a new therapeutic principle (70). In migraine patients without aura, CGRP induced both an immediate and delayed headache with a similar time profile as after GTN and histamine infusion. The induced headaches were milder and the duration of the decreased velocity much shorter than after GTN (51). Interactions or cross-talk exist between NO and CGRP, but the mechanisms remain to be established. In some studies GTN/NO releases CGRP (8,45,92,105); in other studies CGRP activates NOS (7) although some studies show no interactions (20,35,54; see Chapter 16).
NITRIC OXIDE DURING SPONTANOUS MIGRAINE ATTACKS
NO production may be elicited by pathologic reactions such as spreading depression of Leao, the proposed mechanisms of migraine aura (31,81). Increased levels of CGRP were measured during spontaneous migraine headache attacks (28). Sarchielli et al. measured CGRP, nitrites, cGMP, cyclic adenosine monophosphate (cAMP), neurokinin A (NKA), prostaglandin E2 (PGE2) and 6-keto PGF1α (the stable product of PGI2) in serial blood samples obtained from the internal jugular vein throughout a migraine attack (87). An immediate increase in nitrite, cGMP, CGRP, and NKA was observed to indicate activation of the NO and trigeminal systems. Two hours from attack onset PGE2, 6-keto PGF1α, and cAMP also increased; a stress response was excluded. NO is involved in inflammation (22,86) and inhibition of platelet aggregation and adhesion (for review see Radomski et al. [78]). During migraine attacks, changes occur in NO of platelets (15,25,90), and serotonin and NO levels in monocytes (59,92). A rise excitatory amino acids was demonstrated in platelets and plasma (11,21,79), and total thiols were reduced (2).
Related posts:
 Principles of Clinical Pharmacology, Randomized Controlled Clinical Trials, and Evidence-Based Medicine in Headache
Principles of Clinical Pharmacology, Randomized Controlled Clinical Trials, and Evidence-Based Medicine in Headache
 Nitric Oxide
Nitric Oxide
 Human Models of the Headaches
Human Models of the Headaches
 Channelopathies and Their Possible Relation to Migraines
Channelopathies and Their Possible Relation to Migraines
 Autonomic Dysfunction in Migraines
Autonomic Dysfunction in Migraines
 Psychological and Behavioral Treatments of Migraines
Psychological and Behavioral Treatments of Migraines

Full access? Get Clinical Tree


