37 Neuromuscular Disorders in the ICU
 The Motor Unit and Its Physiology
The Motor Unit and Its Physiology Muscles of Respiration
Muscles of Respiration
Three muscle groups may be defined based on their importance for respiration (Figure 37-1):1
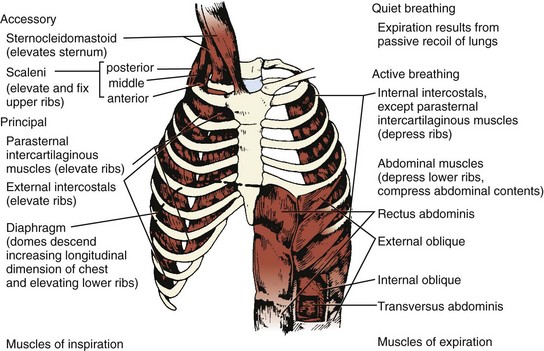
Figure 37-1 Major respiratory muscles.
Inspiratory muscles are indicated on the left and expiratory muscles are indicated on the right.
(From Garrity ER. Respiratory failure due to disorders of the chest wall and respiratory muscles. In: MacDonnell KF, Fahey PJ, Segal MS, editors. Respiratory Intensive Care. Boston: Little, Brown; 1987, p. 313.)
Clinical Presentation of Neuromuscular Respiratory Failure
Patients experiencing respiratory dysfunction due to neuromuscular disease typically present with a combination of upper airway dysfunction and diminished tidal volume (VT). Difficulty with swallowing liquids, including respiratory secretions, is the most typical presentation of pharyngeal weakness, although some patients have an equal or greater degree of difficulty with solid food. A hoarse or nasal voice may also signal problems with the upper airway. These conditions are noted in patients who are at risk for aspiration and present with difficulty with attempts at negative-pressure ventilation (cuirass or iron lung), because the weakened muscles may not be able to keep the airway open as the pressure falls.2 Paradoxical abdominal movement (inward movement of the abdomen during inspiration) is an important sign of diaphragmatic weakness.3
Regardless of the vital capacity, however, indications for intubation and mechanical ventilation include evidence of fatigue, hypoxemia despite supplemental oxygen administration, difficulty with secretions, and a rising PaCO2. In the absence of hypercapnia, occasional patients (e.g., those with myasthenia gravis) can be managed under very close observation in an ICU with less invasive techniques (e.g., bilevel positive airway pressure [BiPAP]).4
In addition to vital capacity, trended measurements of the maximum inspiratory pressure (PImax, more typically recorded as negative inspiratory force [NIF]), are useful indicators of ventilatory capacity. Inability to maintain a PImax greater than 20 to 25 cm H2O usually indicates a need for mechanical ventilation. Although the maximum expiratory pressure (PEmax) is a more sensitive indicator of weakness,5 it has not proved to be as useful as an indicator of the need for mechanical ventilation. A more detailed discussion of these variables and their use may be found elsewhere.6,7
Because a patient with neuromuscular respiratory failure has intact ventilatory drive,8 the fall in VT is initially matched by an increase in respiratory rate, keeping the PaCO2 normal or low until the vital capacity becomes dangerously reduced. Many patients initially maintain their PaCO2 in the range of 35 mm Hg because of either (1) a subjective sense of dyspnea at low VT or (2) hypoxia from atelectasis and increasing dead space. When the PaCO2 begins to rise in this circumstance, abrupt respiratory failure may be imminent.
The modest degree of hypoxia in most of these patients worsens when the PaCO2 begins to rise, displacing more oxygen from the alveolar gas. However, aspiration pneumonia and pulmonary embolism are also frequent causes of hypoxemia in these patients. To determine the relative contributions of these conditions to a patient’s hypoxemia, one can use a simplified version of the alveolar gas equation as follows (derived elsewhere)6,7:
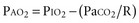
where PAO2 is the alveolar partial pressure of oxygen, PIO2 is the partial pressure of inspired oxygen (in room air, 150 mm Hg), and R is the respiratory quotient (on most diets, about 0.8). This allows estimation of the alveolar-arterial oxygen difference (PAO2 − PaO2). Under ideal circumstances in young people breathing room air, this value is about 10 mm Hg, but it rises to about 100 mm Hg when the fraction of inspired oxygen (FIO2) is 1.0. The alveolar air equation allows one to factor out the contribution of hypercarbia to the decrease in arterial partial pressure of oxygen (PaO2); it should be used to determine whether there is a cause of significant hypoxemia in addition to the displacement of oxygen by carbon dioxide.
Patients with orbicularis oris weakness may have artifactually low vital capacity and NIF measurements because they cannot form a tight seal around the spirometer mouthpiece. The need for nursing and respiratory therapy personnel who are experienced in the care of these patients is thus underscored. It is also important for physicians to observe these patients directly rather than relying solely on reported measurements. The physical findings associated with neuromuscular respiratory failure are reviewed elsewhere.6,7 Among the most important findings are rapid, shallow breathing,9 the recruitment of accessory muscles, and paradoxical movement of the abdomen during the respiratory cycle. Fluoroscopy of the diaphragm is occasionally valuable for the diagnosis of diaphragmatic dysfunction.10
Autonomic dysfunction commonly accompanies some of the neuromuscular disorders requiring critical care, such as Guillain-Barré syndrome, botulism, and porphyria (Table 37-1). In Guillain-Barré syndrome (discussed later) dysautonomia is common and may arise in parallel with weakness or may follow the onset of the motor disorder after one week or more.
TABLE37-1 Neuromuscular Causes of Acute Respiratory Failure
| Location | Disorder | Associated Autonomic Dysfunction? |
|---|---|---|
| Spinal cord | Tetanus112 | Frequent |
| Anterior horn cell | Amyotrophic lateral sclerosis113 | No |
| Poliomyelitis | No | |
| Rabies | Frequent | |
| West Nile virus flaccid paralysis | No | |
| Peripheral nerve | Guillain-Barré syndrome | Frequent |
| Critical illness polyneuropathy | No | |
| Diphtheria | No, but cardiomyopathy and arrhythmias may occur | |
| Porphyria | Occasional | |
| Ciguatoxin (ciguatera poisoning) | Occasional | |
| Saxitoxin (paralytic shellfish poisoning) | No | |
| Tetrodotoxin (pufferfish poisoning) | No | |
| Thallium intoxication | No | |
| Arsenic intoxication114,115 | No | |
| Lead intoxication | No | |
| Buckthorn neuropathy | No | |
| Neuromuscular junction | Myasthenia gravis | No |
| Botulism116 | Frequent | |
| Lambert-Eaton myasthenic syndrome117 | Yes, frequent dry mouth and postural hypotension | |
| Hypermagnesemia118 | No | |
| Organophosphate poisoning | No | |
| Tick paralysis | No | |
| Snake bite | No | |
| Muscle | Polymyositis/dermatomyositis | No |
| Acute quadriplegic myopathy | No | |
| Eosinophilia-myalgia syndrome119 | No | |
| Muscular dystrophies120 | No, but cardiac rhythm disturbances may occur | |
| Carnitine palmitoyl transferase deficiency | No | |
| Nemaline myopathy121 | No | |
| Acid maltase deficiency122 | No | |
| Mitochondrial myopathy123 | No | |
| Acute hypokalemic paralysis | No | |
| Stonefish myotoxin poisoning | No | |
| Rhabdomyolysis | No | |
| Hypophosphatemia124 | No |
 Neuromuscular Disorders
Neuromuscular Disorders
Many chronic neuromuscular disorders and other central nervous system conditions affecting the suprasegmental innervation and control of respiratory muscles eventually compromise ventilation. In this chapter, however, we emphasize the more common acute and subacute neuromuscular disorders that precipitate or prolong critical illness due to ventilatory failure and autonomic dysfunction. A more complete listing of neuromuscular diseases appears in Table 37-1; reviews of this subject11,12 or the references listed in Table 37-1 may be consulted for details of the more rare disorders. Some of the diseases listed (e.g., Lambert-Eaton myasthenic syndrome) rarely cause respiratory failure in isolation but may be contributing causes in the presence of other conditions13 such as neuromuscular junction blockade intended only for the duration of a surgical procedure.14
Neuromuscular Diseases Precipitating Critical Illness
Guillain-Barré Syndrome
Guillain-Barré syndrome, or acute inflammatory demyelinating polyradiculoneuropathy, is typically a motor greater than sensory peripheral neuropathy with subacute onset, monophasic course, and nadir within 4 weeks. Although the precise etiology is unknown, Guillain-Barré syndrome is immune mediated and related to antibodies directed against peripheral nerve components. Approximately 1.7 cases occur per 100,000 population per year.15 Most patients suffer a demyelinating neuropathy, but in about 5% of cases the condition is a primary axonopathy.16 Numerous antecedents have been implicated17; the more frequent ones are listed in Box 37-1. The association with antecedent infections suggests that certain agents may elicit immune responses involving antibodies that cross-react with peripheral nerve gangliosides. In particular, the development of ganglioside antibodies has been observed in Guillain-Barré syndrome after Campylobacter jejuni infections, such as GM1 antibodies in axonal forms of Guillain-Barré syndrome18 and GQ1b antibodies in the Miller-Fisher variant of Guillain-Barré syndrome.19
The initial findings of patients with Guillain-Barré syndrome are subacute and progressive weakness, usually most marked in the legs, associated with sensory complaints but without objective signs of sensory dysfunction.20 Deep tendon reflexes are often significantly reduced or absent at presentation, though this finding may take several days to develop. The cerebrospinal fluid (CSF) typically reveals an albuminocytologic dissociation or elevated protein content without pleocytosis; this may not evolve until the second week of illness. The major reason to examine the CSF is to preclude other diagnoses. Although mild CSF lymphocytic pleocytosis (10-20 cells/mm3) may suggest the possibility of associated human immunodeficiency virus (HIV) infection, in most patients, the nucleated cell count is less than 10 cells/mm3.21 Although they may be normal initially, results of electrodiagnostic studies (motor and sensory nerve conduction studies and needle electromyography) often reflect segmental nerve demyelination with multifocal conduction blocks, temporally dispersed compound muscle action potentials, slowed conduction velocity, and prolonged or absent F waves.22 Differential diagnostic considerations for patients with suspected Guillain-Barré syndrome are primarily those listed in the “Peripheral Nerve” section of Table 37-1.
The components of treatment for patients with Guillain-Barré syndrome are as follows:
Many patients are too weak to trigger the ventilator; in such cases, the assist/control or intermittent mandatory ventilation mode is initiated. Weaning patients with Guillain-Barré syndrome from mechanical ventilation must wait for adequate improvement in strength. We usually shift to pressure support ventilation for weaning, although evidence of its superiority over intermittent mandatory ventilation or synchronized intermittent mandatory ventilation modes is only anecdotal. Although the majority of patients require mechanical ventilation for less than 4 weeks, as many as one-fifth need 2 or more months of support before they can breathe without assistance. Improvement in vital capacity to greater than 15 mL/kg and in NIF to greater than 25 cm H2O suggests that a patient has improved enough to begin weaning from the ventilator. A formula using a combination of ventilatory and gas exchange variables may allow more accurate determination of a patient’s ability to be weaned.23
Nutritional support should begin as soon as a patient is admitted, with appropriate concern for the risk of aspiration.24 Most mechanically ventilated patients with Guillain-Barré syndrome can be fed via soft, small-caliber feeding tubes; autonomic dysfunction affecting the gut occasionally requires total parenteral nutrition.
Immunotherapy for Guillain-Barré syndrome includes removal of autoantibodies with plasma exchange or immune modulation with high-dose intravenous immunoglobulin (IVIg). The efficacy of plasma exchange has been evaluated in a Cochrane systematic review of six class II trials comparing plasma exchange alone with supportive care.25 Most of the trials employed up to 5 plasma exchanges of 50 mL/kg over 2 weeks. In a large North American trial,25 the time needed to improve one clinical grade (being weaned from the ventilator or being able to walk) was reduced by 50% in the plasma exchange group by comparison with the control group. There was no significant benefit when plasma exchange was begun later than 2 weeks after symptom onset. A meta-analysis demonstrated more rapid recovery in ventilated patients treated with plasma exchange within 4 weeks of onset.26 The optimal number of plasma exchanges has been assessed in patients with mild (unable to run), moderate (unable to stand without assistance), and severe (requiring mechanical ventilation) Guillain-Barré syndrome by the French Cooperative Group.27 On the basis of this trial, two exchanges are better than none in mild Guillain-Barré syndrome; four are better than two in moderate Guillain-Barré syndrome; and six are no better than four in severe Guillain-Barré syndrome. Albumin is the preferred replacement solution.28 Treatment with IVIg for Guillain-Barré syndrome has also been examined in a Cochrane systematic review. Three randomized controlled trials demonstrated class I evidence that IVIg (2 g/kg over 2-5 days) is as effective as plasma exchange in Guillain-Barré syndrome patients with impaired walking.29 Complication rates were somewhat higher in the plasma exchange groups. A large international multicenter randomized trial compared plasma exchange (50 mL/kg × 5 exchanges over 8-13 days), IVIg (0.4 g/kg × 5 days), and plasma exchange followed by IVIg.30 No significant outcome differences between these therapies were found with respect to functional improvement at 4 weeks or at 48 weeks.
Evidence-based guidelines for Guillain-Barré syndrome immunotherapy have been published by the Quality Standards Subcommittee of the American Academy of Neurology.31 Plasma exchange is recommended for adult patients who cannot walk within 4 weeks of symptom onset. IVIg is recommended in these patients within 2 or possibly 4 weeks of symptom onset. Both treatments are deemed equivalent in efficacy, and combining treatment with plasma exchange and IVIg confers no additional benefit. In light of their therapeutic equivalence, the decision whether to employ plasma exchange or IVIg in treating acute Guillain-Barré syndrome may be determined by resource availability and by avoiding potential side effects related to a patient’s medical comorbidities. Patients with heart disease, renal insufficiency or failure, hyperviscosity, or IgA deficiency may be more susceptible to complications of treatment with IVIg, whereas plasma exchange may be complicated in patients with labile blood pressure, septicemia, and significant venous access problems.
Despite the autoimmune pathophysiology of Guillain-Barré syndrome and the efficacy of corticosteroids in more chronic forms of inflammatory neuropathy, corticosteroids have not demonstrated effectiveness in Guillain-Barré syndrome and are therefore not recommended for Guillain-Barré syndrome treatment.31 A large multicenter trial failed to demonstrate efficacy of high-dose intravenous methylprednisolone,32 and another large multicenter trial demonstrated no added clinical benefit in combined treatment with IVIg and methylprednisolone.33

Full access? Get Clinical Tree


