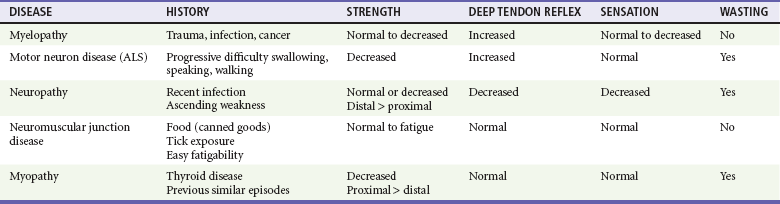
Chapter 108 The neuromuscular unit has four components: the anterior horn cells of the spinal cord, the peripheral nerve, the neuromuscular junction, and the muscle innervated. The level of the pathologic process determines associated signs and symptoms (Table 108-1). Myelopathies involve the spinal cord; radiculopathies involve the nerve roots as they leave the spinal cord; neuropathies involve the peripheral nerves; and myopathies involve the muscle. The use of physical signs to differentiate these disorders is discussed in Chapter 13. The assessment of vital signs is important because some causes of weakness may result in dysregulation of the autonomic system. A systematic neurologic examination assesses the patient’s mental status, cranial nerves, motor function, sensory function, deep tendon reflexes, and coordination, including cerebellar function. The motor examination begins by determining whether the weakness is unilateral or bilateral and which muscle groups are involved. Key components of the examination include motor strength, muscle bulk, and presence of fasciculations. Box 108-1 provides the grading system used in motor strength assessment. Table 108-2 provides the findings used to distinguish upper motor neuron from lower motor neuron processes. Myelopathies are spinal cord disorders that are manifested with signs of upper motor neuron dysfunction, such as muscle weakness with increased spinal reflexes, including an extensor plantar reflex (Babinski’s response). Muscle tone ranges from normal to slightly increased, eventually leading to spasticity. There may be bladder and bowel involvement. When sensory findings are present, they often define the level of the lesion. The presence of back pain suggests a compressive lesion, such as a herniated intravertebral disk, epidural hematoma, abscess, or tumor. Acute, painless spinal cord lesions include transverse myelitis and spinal cord infarction. Myelopathies are discussed in Chapter 106. Weakness from a neuropathy is often noted first in distal muscles and then ascends. Decreased grip strength and footdrop are common presentations. Muscle tone ranges from slightly diminished to flaccid, and deep tendon reflexes are diminished or absent. Patients exhibit varying degrees of altered sensation, muscle wasting, and fasciculations, depending on the duration of the symptoms. Disorders in the differential diagnosis include GBS, toxic neuropathies, diabetic neuropathy, and tick paralysis (which is caused by inhibition of both nerve conduction and function of the neuromuscular junction). Neuropathies are discussed in Chapter 107. Disorders of the Neuromuscular Junction Perspective.: MG affects approximately 60,000 Americans.1 The age at onset is bimodal; women are most commonly affected between the ages of 20 and 40 years and men between 50 and 70 years. Whereas new cases of MG are occasionally diagnosed in the emergency department (ED), it is much more common for patients with established disease to present with exacerbations of their disorder, often caused by precipitating factors. Principles of Disease.: MG results from autoantibodies directed against the nicotinic AChR at the neuromuscular junction or from antibodies against muscle-specific tyrosine kinase. This leads to destruction of AChRs with a decrease in the total number of available receptors. The autoantibodies further compete with ACh for binding at remaining receptors. With repeated stimulation, fewer and fewer receptor sites are available for ACh binding, and fatigue develops. Clinical Features.: Patients with MG present with easy fatigability as the result of progressive weakness with repeated activity of affected muscle groups. Ocular symptoms are often the first manifestation of MG; typical symptoms are ptosis, diplopia, and blurred vision. Ocular muscle weakness is the first sign in up to 40% of patients, although 85% of patients with MG eventually have ocular involvement. When ptosis is present, it is often worse toward the end of the day. Respiratory failure is rarely the initial symptom of MG. Even so, up to 17% of patients may have weakness of the muscles of respiration.2 Bulbar muscles may be involved, producing dysarthria or dysphagia. Lambert-Eaton myasthenic syndrome is a rare disorder. Almost 50% of cases are associated with small cell carcinoma of the lung. Autoantibodies cause inadequate release of ACh from nerve terminals, affecting both nicotinic and muscarinic receptors. With repeated stimulation, the amount of ACh in the synaptic cleft increases, leading to an increase in strength, the opposite of that seen with MG. The classic syndrome includes weakness that improves with use of muscles, particularly proximal hip and shoulder muscles; hyporeflexia; and autonomic dysfunction, most commonly seen as dry mouth.3 Management primarily focuses on treatment of the underlying neoplastic disorder, although intravenous immune globulin (IVIG) has been reported to be useful.4
Neuromuscular Disorders
Principles of Disease
Clinical Findings
Physical Examination
Differential Consideration
Neuropathies
Specific Disorders
Myasthenia Gravis

Full access? Get Clinical Tree


Neuromuscular Disorders

Only gold members can continue reading. Log In or Register to continue




