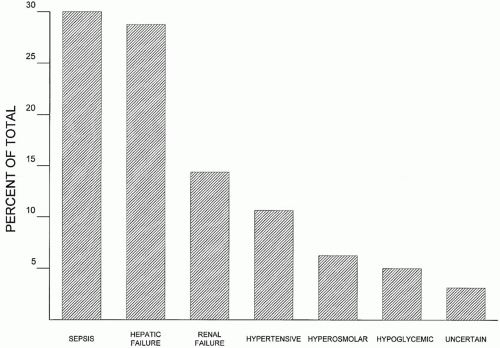
 ▪ FIGURE 34-1 Relative frequency of (nonsedative) causes of delirium in nontrauma ICU patients. Sepsis, hepatic failure, and renal failure rank as the most common causes. |
Removal of unnecessary tubes and monitoring devices and beginning a program of physical activity as soon as feasible may also serve to prevent or reverse delirium. Even if none of these measures are beneficial for delirium, they will not be harmful and may offer noncognitive therapeutic benefits.
TABLE 34-1 CLINICAL CHARACTERISTICS OF CAUSES OF COMA | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
hypoglycemia, dehydration, sepsis, hepatic encephalopathy, and uremia can all contribute to impaired consciousness. The common reversible causes of coma are summarized in Table 34-2.
TABLE 34-2 COMMON REVERSIBLE CAUSES OF ALTERED MENTAL STATUS | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
is the finding that inflammatory cytokines (e.g., interleukin-1) may directly impair consciousness. Sepsis-induced coma is particularly common in the febrile elderly patient, where altered mental status presents a difficult management decision. Frequently deliberated diagnoses are severe sepsis alone, or sepsis complicated by dehydration, electrolyte abnormalities, meningitis, and intracranial structural lesions such as a brain abscess or hemorrhage. Timely antibiotic therapy is desirable in all infectious situations, but confirming the diagnosis and nailing down a specific organism so as to best choose antimicrobial therapy usually requires a lumbar puncture (LP). Unfortunately, LP incurs some risk in the presence of coagulopathy, anticoagulation, or intracranial mass lesion. Therefore, deciding the order in which to perform a head computed tomography (CT) scan, do an LP, and administer antibiotic therapy is often debated. In febrile nonimmunocompromised patients without evidence of a focal neurologic defect, papilledema, or history of trauma, an LP is almost certainly safe and can be performed before or without a head CT scan. (Not that long ago, the decision to perform an LP was always made only based upon history and examination.) If head trauma, papilledema, new onset seizures, or focal neurologic deficit complicates the evaluation of a potentially infected patient, probably the best course is to obtain blood cultures, administer empiric antibiotics, and proceed to the CT scanner before LP. In any case, if logistical limitations preclude prompt CT scanning and intracranial infection is a real possibility, it is best to begin antibiotic treatment, even if doing so precludes a definitive microbiological diagnosis. Although it is rational to administer antibiotics as quickly as practical in patients with suspected meningitis, there is little if any data to support the often-quoted need to dose antibiotics within 1 h of suspecting the diagnosis.
cause coma, very rapid loss of consciousness without rostrocaudal progression is typical.
considered in patients with altered mental status and hypertension. Cocaine-induced hypertension alone may cause delirium but if coma is present, intracranial hemorrhage is likely. Although young patients without underlying vascular or cerebral disease may remain awake with very low (40 to 50 mm Hg) MAPs, the elderly, patients with chronic hypertension, and those with concurrent metabolic encephalopathy or structural lesions tolerate hypotension less well.
nonfunctional. If only the eye ipsilateral to caloric stimulus abducts, a lesion of the medial longitudinal fasciculus (encapsulated by the pons) should be suspected.
TABLE 34-3 THE GLASGOW COMA SCALE | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
with obvious head trauma, because of the possibility that a metabolic cause may coexist or may have precipitated the trauma (e.g., alcohol, carbon monoxide). Laboratory determinations should include indices of renal and hepatic function, serum glucose and electrolyte determinations, hemoglobin and arterial blood gases, and when appropriate, carboxyhemoglobin determinations. If the history, physical examination, or initial laboratory testing suggests drug overdose or poisoning, a toxicology profile, and if indicated specific levels of compounds not included in a typical toxicology screen (e.g., aspirin, acetaminophen, ethylene glycol, methanol), should be obtained (see Chapter 33).
recurrence; however, it should be remembered that even patients with epilepsy develop strokes, tumors, and CNS infections. Hence, recurrent seizures should not be reflexly ascribed to a singular cause in perpetuity.
trial comparing lorazepam to phenytoin, diazepam plus phenytoin, or phenobarbital, lorazepam alone was the most effective initial therapy. A minor disadvantage is the need to refrigerate lorazepam. Diazepam (0.15 mg/kg) is an acceptable alternative that does not require refrigeration. Diazepam is also available as a rectal gel that can be used when IV dosing is not possible. The major disadvantages of diazepam are its shorter duration of action and potent scleroscent effect on peripheral veins. Midazolam (0.2 mg/kg bolus) can also be used in place of lorazepam, but because it has the shortest duration of action of all the benzodiazepines, a continuous infusion (0.2 to 1 μg/kg/min) is usually required. Hypotension and hypopnea are complications of all benzodiazepines but occur rarely (<5%) unless other anticonvulsants (especially, phenobarbital) have been administered.
TABLE 34-4 THERAPY OF STATUS EPILEPTICUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:







Full access? Get Clinical Tree


