Chapter 102 Neuroendocrine–Immune Mediator Coordination and Disarray in Critical Illness
Organization of the Stress Response
Stress can be understood as any threat to an organism’s homeostatic internal milieu. An organism must defend itself against two types of threats. Macroscopic threats, such as an encounter with a predator, an episode of hypotension, or a traumatic hemorrhage, threaten the whole organism at once. The central nervous system response to macroscopic threats has been recognized for decades. Microscopic threats attack epithelial and endothelial barriers that are essential to organ function. To maintain integrity against microscopic threats, the organism must be vigilant against microbial invasion in the face of the wear and tear of daily activities such as digestion, excretion, and procreation. The central stress response to macroscopic threats puts housekeeping on hold as the vital functions are marshaled against an assault. The response to microscopic threats is a matter of day-to-day housekeeping and is primarily the responsibility of the immune system. They are equally important and must balance each other; they are the yin and the yang of the response to stress.
Central Stress Response
All types of stress—physical, emotional, or immune—cause stimulation of the sympathetic nervous system, activation of the HPA axis, and release of hypothalamic hormones, including vasopressin, prolactin, and growth hormone (Figure 102-1). The HPA system is the major humoral coordinator of the central stress response (see Chapter 77). Hypothalamic release of corticotropin-releasing hormone occurs in response to cortical signals generated by fear, pain, or hypotension, as well as in response to immune-derived signal molecules, especially interleukin (IL-1β), tumor necrosis factor (TNF-α), and IL-6.1 Corticotropin-releasing hormone in the brain stimulates sympathetic outflow and causes release of adrenocorticotropic hormone (ACTH) by the pituitary. Vasopressin, that is also released in response to immune signals and stress, is powerfully synergistic with corticotropin-releasing hormone in promoting ACTH secretion.2 Circulating ACTH reaches the adrenal cortex, where it stimulates conversion of cholesterol to the steroid hormone cortisol. Cortisol release suppresses further ACTH secretion within minutes and more slowly feeds back to suppress secretion of corticotropin-releasing hormone.3 Cortisol and its synthetic analogues, the glucocorticoids, act in tandem with the sympathetic nervous system to prepare a body for action by putting growth and housekeeping functions on hold, making fuel substrates available, and supporting blood pressure and intravascular volume. Failure of this normal stress response leads to cardiovascular collapse.
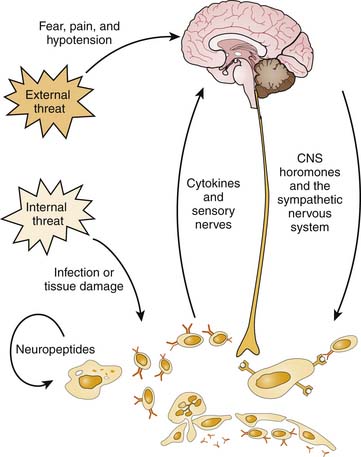
Cortisol inhibits growth in all tissues, decreases deoxyribonucleic acid and ribonucleic acid synthesis, and increases protein catabolism, except in the liver, where protein synthesis is enhanced. Cortisol increases the availability of glucose, amino acids, and free fatty acids for immediate use. Cortisol increases vascular tone and increases expression of adrenergic receptors, potentiating the vasoconstrictive action of catecholamines. Cortisol also regulates the distribution of total body water. It activates the renin-angiotensin system, causing hypertension, and is necessary for secretion of a water load.3 In suppressing the body’s maintenance functions, cortisol suppresses immune responses. Cortisol reduces circulating numbers of lymphocytes, monocytes, and eosinophils by stimulating apoptosis. It stabilizes lysosomal membranes, decreases capillary permeability, impairs demargination of white blood cells and phagocytosis, and decreases release of IL-1, preventing fever. Cortisol supports a TH2 (humoral immunity) over a TH1 (cellular immunity) phenotype.3
Activation of the sympathetic nervous system complements the actions of cortisol. The impact of sympathetic nervous system stimulation on the immune system is complex and imperfectly understood and is discussed later in this chapter. The parasympathetic limb of the autonomic nervous system plays an important role in regulating the immune response. Fibers from the vagus nerve innervate liver macrophages, called Kupffer cells, as well as other abdominal lymphoid organs. Efferent signaling from the vagus nerve decreases the production of inflammatory cytokines by Kupffer cells, macrophages, and other cytokine producing cells, an effect that has been termed the cholinergic antiinflammatory pathway.4 This pathway may be a protective mechanism that blunts the response to sterile endotoxin translocation from the gut. Alternatively, parasympathetic activation may complement the stress response in restraining the magnitude of an inflammatory response to real infection.
Prolactin and growth hormone share structural homology with each other and with the immune-derived cytokines granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-2, and IL-7.5,6 Together these hormones support immune response by stimulating cytokine expression, antibody production, and clonal expansion of lymphocytes. Prolactin and growth hormone directly oppose the lymphocyte apoptosis promoted by cortisol. Growth hormone also affects metabolism, which may be important in critical illness. The overall impact of vasopressin on the immune system is less well understood, but its function in direct interaction with lymphocytes appears to be supportive.
Immune Response to Microscopic Threats
Inflammation and phagocytic cells comprise the first line of defense and recognition. Tissue damage caused by microbial invasion or trauma initiates the nonspecific cascade called the inflammatory response (see Chapters 90 and 91). It leads to vasodilatation, increased capillary permeability, and an influx of phagocytes. Histamine and kinins are important mediators of the inflammatory response. Phagocytic cells may be blood monocytes, neutrophils, or tissue macrophages. Macrophages engulf foreign matter and bacteria, break them down into large molecules, and present these as antigens to T lymphocytes, thus activating the specific adaptive immune response.
Phagocytic cells also produce proinflammatory cytokines, primarily IL-1, IL-6, and TNF-α, which amplify the immune response locally. Cytokines help determine whether the specific immune response takes on a TH1 or TH2 phenotype. Cytokines carry messages between different cell types within the immune system and between the immune system and other organ systems. In the latter role, they are thought to comprise the most important afferent limb of the NEI system. Cytokines circulating in the bloodstream reach the central nervous system, where receptors for IL-1α and IL-1β, IL-2, IL-4, IL-6, TNF-α, and interferon (IFN-γ) are found. Afferent nerves also carry inflammatory signals to the brain. When stimulated by cytokines and endotoxin, signals from the vagus lead to HPA axis activation.7
Interestingly, neuroendocrine mediators that trigger the antiinflammatory cascade in the central nervous system, such as vasopressin and corticotropin-releasing hormone, may have the opposite effect when they are released in tissues. Both corticotropin-releasing hormone and vasopressin are clearly immunosupportive in peripheral tissues. Corticotropin-releasing hormone is found in high concentrations at inflammatory sites, although plasma levels may be undetectable.8,9 Corticotropin-releasing hormone augments T-lymphocyte proliferation and IL-2 expression in vitro, augments cytokine expression by macrophages, and triggers mast cell degranulation.8,10 Immunoneutralization of corticotropin-releasing hormone decreases inflammation as effectively as neutralizing antibodies to TNF-α.
Acute Versus Chronic Stress
The body’s response to stress has been studied mostly as an acute event occurring in normal healthy people. Through the use of advanced therapies and machines, we have created in the modern ICU a state of prolonged critical illness that does not exist in nature and to which the neuroendocrine system must adapt. The state of prolonged stress is not well understood, but the hormonal milieu that exists and which we intentionally create in the acute phase of resuscitation—that of catecholamine and cortisol excess—likely will not be appropriate later. In chronic critical illness, the excess of ACTH, prolactin, growth hormone, and thyroid hormone observed initially is reversed, and the normal pulsatile pattern of pituitary hormones is diminished or lost.11 It is unclear whether this represents a beneficial adaptation to critical illness or an unrecognized organ failure.
Neuroendocrine–Immune Dysfunction Causing Critical Illness
Hypothalamic-Pituitary-Adrenal Axis
In general, critically ill patients have elevated cortisol levels that correspond to the severity of illness. In the acute phase of critical illness, adequate cortisol is necessary for maintenance of vascular tone and normal catecholamine responsiveness.12 In some critically ill patients, cortisol levels may be inadequate for their degree of illness or inadequate relative to the ACTH levels measured in their blood. The incidence of relative adrenal insufficiency varies according to author and definition criteria. The reported incidence ranges from 0% to 75% and is higher in patients with septic shock than other ICU patients. Relative adrenal insufficiency is associated with poor outcomes in adults and children.13,14
During the prolonged phase of critical illness, the normal association between ACTH and cortisol often is strikingly reversed. In the face of ongoing stress, cortisol levels in critically ill patients tend to remain elevated despite a decline in ACTH levels.15 Sustained cortisol production may result from direct autonomic innervation of steroidogenic cells, paracrine action of the products of the adrenal medulla, or the action of cytokines, which can directly stimulate cortisol production.16–18 Decreased cortisol clearance by the liver may contribute to elevated cortisol levels in prolonged critical illness.15
Adrenal insufficiency should be suspected in any patient with catecholamine-resistant shock and in patients with prolonged critical illness. Pathologic scenarios that may produce relative or absolute adrenal insufficiency include suppression of corticotropin-releasing hormone or ACTH secretion, adrenal unresponsiveness, increased cortisol clearance, or end-organ unresponsiveness to cortisol (Table 102-1). For a thorough discussion of the causes of relative adrenal insufficiency, the reader is referred to the excellent review by Beishuizen and Thijs.19
Table 102–1 Causes of Adrenal Insufficiency
| Nervous System | Condition | Comments |
|---|---|---|
| Central | Hypothalamic or pituitary disease | |
| Brain injury | ||
| Recent steroid use | Follows the “rule of ones”: 1 day of steroid use may suppress adrenal function for 1 week; 1 week for 1 month; 1 month for 1 year | |
| Peripheral | Preexisting adrenal failure | Associated with increased pigmentation, hypoglycemia, mild hyponatremia, and hyperkalemia |
| Acute adrenal failure | Adrenal hemorrhage or autoimmune adrenalitis | |
| Inadequate substrate | Low cholesterol48 | |
| P450 impairment | From ketoconazole, etomidate, sepsis, prematurity, or age <6 months77–79 | |
| Increased clearance | Occurs with rifampin, phenytoin, and phenobarbital80 | |
| End-organ unresponsiveness | Cytokines can change glucocorticoid receptor sensitivity19,80 | |
| Other | Sepsis/inflammation | Circulating inflammatory mediators can cause hypothalamic-pituitary-adrenal axis suppression, particularly in patients with fungal infection or human immunodeficiency virus18 |
Defining what constitutes a normal cortisol response to critical illness is difficult because the range of cortisol levels observed in critically ill patients is so broad, varying from the healthy normal level to 20 times normal. Additionally, measured cortisol levels may overestimate biologically active free cortisol in the acute phase of critical illness because levels of cortisol-binding globulin are low.20,21 Most authors agree that a “normal” level of cortisol in the face of shock probably is inadequate.19
The ACTH stimulation test can be used to identify absolute adrenal failure but may fail to identify a clinically significant adrenal insufficiency. The ACTH dose used in the standard test (250 mg for adults and children older than 2 years, 125 mg for children younger than 2 years) produces plasma ACTH levels 100 to 200 times the physiologic maximum stress levels of ACTH. A 1-mg test has been suggested but has not been standardized for adults or children. Beishuizen et al.22 demonstrated that patients with a normal response to the standard test but an inadequate response to the 1-mg test improved clinically with decreased pressor requirement in response to hydrocortisone treatment. In adult patients with septic shock, a random cortisol level less than 25 μg/dL was a better predictor of a hemodynamic response to hydrocortisone than either the 1-mg or the 250-mg ACTH stimulation test.23 Most recently, in a study of 30 children with septic shock, a change in cortisol of greater than 9 μg/dL in response to a 1 mg ACTH stimulation test identified patients at increased risk of catecholamine refractory shock but not increased risk of mortality.24
Standard reference ranges for normal healthy subjects accept minimum peak values after ACTH stimulation of 18 to 22 mg/dL (500-600 nmol/L) or a minimum change in cortisol level of 7 to 9 mg/dL (200-250 nmol/L). Failure according to these requirements is associated with a high 28-day mortality in adults.13 ACTH stimulation tests should be interpreted in light of the basal cortisol level, because the change in serum cortisol after ACTH stimulation is a measure of adrenal reserve.
It may be impossible to rely on results of an ACTH stimulation test in the acute setting of severe catecholamine-unresponsive shock, when time is of the essence. Because the criteria for diagnosis of relative adrenal insufficiency in the critical care setting are imprecise, some authors believe a negative or inconclusive result should not prohibit use of glucocorticoids. Instead, a rapid hemodynamic response to glucocorticoids may be the best clue to the diagnosis of relative adrenal insufficiency. One author suggests that, at present, we have no choice but to rely on clinical assessment of the severity of stress and make “an educated guess of the adequacy of the measured serum cortisol concentration.”21
Although several studies on administration of pure glucocorticoids to septic shock patients have failed to demonstrate a survival advantage, the use of steroids with both glucocorticoid and mineralocorticoid activity has shown some benefit.26,27 Annane et al.25 reported a large, prospective, placebo-controlled, randomized clinical trial in adults with septic shock, demonstrating that treatment with hydrocortisone (50 mg every 6 hours) and fludrocortisone (50 mg/day) decreased 28-day mortality in all patients. Most of the survival benefit was seen in patients in whom relative adrenal insufficiency was identified by standard-dose corticotropin stimulation test with change in stimulated minus baseline cortisol response of less than 9 μg/dL.25 A large, international, multicenter trial (CORTICUS trial) investigating this effect found that hydrocortisone given without fludrocortisone hastened resolution of shock but had no impact on mortality.28 A high percentage of patients in the Annane study were treated with etomidate, an inhibitor of the cytochrome P-450 enzymes responsible for the final step in cortisol synthesis. An a priori subgroup analysis of the patients in the CORTICUS trial who received etomidate for intubation found an increased risk of adrenal insufficiency and mortality in these patients. Treatment with hydrocortisone did not change the mortality of patients who received etomidate.29
A recent meta-analysis of clinical trials of steroids in septic shock concluded that low-dose long-duration corticosteroids may have an advantage over high doses and shorter duration.30 Current recommendations for adult sepsis recommend use of low-dose steroids (<300 mg/day of hydrocortisone) for shock that is resistant to fluid resuscitation and catecholamines, without use of the ACTH stimulation test.31,32 An excellent historical review of the use of steroids for septic shock reminds us that there is still need for clinical equipoise on the use of steroids for septic shock in children.33 At present, the American College of Critical Care Medicine clinical practice parameters for the hemodynamic support of pediatric and neonatal septic shock recommend the use of hydrocortisone in patients with catecholamine resistance and suspected adrenal insufficiency. Risk factors that should alert the clinician to high risk for adrenal insufficiency include physical examination findings suggestive of absolute adrenal insufficiency, disseminated intravascular coagulation or purpura fulminans, and history of steroid use. The authors would add to this list a history of prolonged stress (i.e., decompensation in a patient during a long ICU stay), severe septic shock, and risk of P-450 impairment as a result of drugs and extreme youth.
The physiologic replacement dose of hydrocortisone is 12.5 mg/m2/day. In critical illness, dosing ranges from 50 mg/m2/day in stress states to 50 mg/kg/day in severe shock have been reported, although dosing greater than 300 mg/day of hydrocortisone equivalent is associated with worse outcomes.31,32 Hydrocortisone has a short half-life and should be given as a bolus, followed by continuous infusion or dosing every 6 hours as per most recent interventional trials. Although critically ill patients may receive steroids for a variety of indications, steroids that lack mineralocorticoid activity may not be sufficient for adrenal replacement in shock. Table 102-2 gives a comparison of the relative glucocorticoid and mineralocorticoid potency of steroids commonly used in the ICU. Glucocorticoids given over a prolonged period are associated with an increased risk for superinfection, gastrointestinal bleeding, hyperglycemia, polyneuropathy of critical illness, and prolonged adrenal insufficiency. Exogenous hydrocortisone should be tapered or discontinued as soon as hemodynamics allow.
Table 102–2 Comparison of Common Corticosteroid Dosing
| Dosage | Glucocorticoid Potency (as Hydrocortisone Equivalent Dose) | Mineralocorticoid Potency |
|---|---|---|
| Stress dose hydrocortisone: 50 mg/m2/day | Approximately 2 mg/kg/day | ++ |
| Shock dose hydrocortisone: 50 mg/kg/day | 50 mg/kg/day | ++ |
| Annane study3: hydrocortisone 50 mg q6h and fludrocortisone 50 mg/day | Approximately. 3 mg/kg/day | ++++ |
| Dexamethasone for postextubation stridor: 0.5 mg/kg q8h | 35 mg/kg/day | 0 |
| Methylprednisolone in severe asthma: 1 mg/kg q6h | 20 mg/kg/day | 0 |
| Methylprednisolone in spinal cord injury: 30 mg/kg load followed by 5.4 mg/kg/h over 23 hours | 770 mg/kg/day | 0 |






Full access? Get Clinical Tree


