
MULTIPLE ORGAN DYSFUNCTION SYNDROME
Multiple organ dysfunction syndrome (MODS) is a potentially progressive syndrome of reversible dysfunction of two or more organ systems as a consequence of acute, life-threatening, pathologic interactions among host defenses, the inflammasome, and the coagulation system that, once deranged, disrupt systemic homeostasis. Nearly 40 years since the initial description,1 MODS is unquestionably the leading cause of death among critically ill surgical patients, and the leading cause of late deaths (not related to exsanguination or overwhelming central nervous system [CNS] injury) following trauma.2 Moreover, the financial burden to both individual hospitals and the health care system as a result of the acute care of patients with MODS is astronomical, and the long-term needs of survivors are substantial.3
Currently, the MODS is believed to represent a severe manifestation of dysregulated or uncontrolled systemic inflammation.4,5 Despite a sophisticated understanding of the pathogenesis of MODS, effective therapies have remained elusive; therefore prevention or early mitigation becomes crucial if lives are to be saved. This is likely because of the heterogeneous manifestations of MODS; organ dysfunction may follow a vast array of physiologic or metabolic insults, and may affect variable numbers of organ systems to varying degrees at different times. The development of targeted therapeutics has been hampered by the redundancy and interrelationships of the dysregulated immune response, or because such therapies have been targeted too narrowly, if not mistargeted altogether.6 Better understanding of these interrelationships is crucial if patient care is to improve or new therapies are to be found.
HISTORICAL PERSPECTIVES
Multiple organ failure (MOF) following a severe physiologic insult was recognized first during the late 1960s as the unwanted consequence of advancements in the management of shock. Successful resuscitation of shock was followed by death from hitherto unidentified disease manifestations characterized by progressive, irreversible failure of several organs. In 1975, Baue coalesced early case reports of organ failure following severe injury7,8 into the concept of MOF as a distinct entity, which he described as “the progressive failure of many or all systems after an overwhelming injury or operation.”9 In doing so, Baue gleaned two concepts fundamental to mortality during critical illness: First, that mortality in the intensive care unit (ICU) was the consequence of the interaction of multiple failing organs; and second, that individual organ systems were interrelated; injury to one organ system could cause dysfunction of another. For example, pulmonary failure was found more often than not to occur along with dysfunction of at least one other organ system.10 Moreover, mortality from acute respiratory failure is usually determined by the magnitude of nonpulmonary organ dysfunction; the combination of respiratory and hepatic dysfunction is especially deleterious.10,11 Furthermore, improvements in the treatment of the acute respiratory distress syndrome (ARDS), such as ventilation at low tidal volumes, also decreased the likelihood of additional, subsequent organ failure.12,13 Soon after Baue’s initial description of MOF, Fry et al.14 reported a linear relationship between the number of failed organs and mortality during critical illness; whereas mortality following failure of a single organ was 30%, mortality following failure of four or more organs was 100%.
The etiology of MOF was believed initially to be always infection.15 However, clinical, pathologic, and experimental findings have discredited this theory. Autopsies of patients with MOF did not always demonstrate a focus of infection; either infection was never present, or organ dysfunction progressed despite successful anti-infective therapy.16,17 Trauma patients with organ failure were sometimes never infected.18 When infection did occur during critical illness, it sometimes followed organ failure, rather than preceding it.19 Under experimental conditions, characteristic hemodynamic and inflammatory derangements could be replicated absent any infection.20,21
This discrepancy was reconciled by observations that an occult reservoir of pathogens (e.g., the gastrointestinal [GI] tract) could initiate or perpetuate sepsis and organ dysfunction without any overt infection.22,23 According to this “gut-motor” hypothesis, bacterial overgrowth during critical illness (secondary to gastric acid suppressive therapy, impaired intestinal immunity, or both) caused organ dysfunction. However, selective gut decontamination (topical oral antibiotics and enteral antibiotics by gavage, with or without systemic antibiotic prophylaxis) although effective in reducing the incidence of nosocomial infection, neither attenuated MODS nor improved mortality, calling the gut-motor hypothesis into question.21,24 Even if ultimately correct, it may be that mediators (elaborated in intestinal lymph as a result of intestinal ischemia-reperfusion (I-R) injury, thence becoming systemic) are more important than the intestinal flora.
That infection was sufficient, but unnecessary to cause organ damage stimulated the reevaluation of the pathophysiology of MOF. Goris et al.18 suggested “massive activation of inflammatory mediators by severe tissue trauma or intra-abdominal sepsis” as the etiology of MOF. Marshall and Sweeney25 reinforced this theory by observing that the degree of the inflammatory response to infection predicted ICU mortality, rather than the type or extent of infection itself. Eventually, the hypothesis developed that a hypodynamic, excessive, or otherwise dysfunctional immune response was the principal cause of organ damage, rather than the cytotoxic effects of invading microorganisms per se.4 This theory synthesized myriad, seemingly unrelated causes of organ failure into a unifying hypothesis, and was consistent with extant clinical and experimental observations. In recognition, and in an attempt at standardization, an American College of Chest Physicians/Society of Critical Care Medicine consensus statement defined diagnostic criteria for what was termed the systemic inflammatory response syndrome (SIRS) in 1992.26 The diagnosis of SIRS was fulfilled by the presence of at least two of the host-response criteria: (1) Core body temperature >38°C or <36°C, (2) heart rate >90 beats/min, (3) respiratory rate >20 breaths/min (not ventilated) or PaCO2 < 32 mm Hg (ventilated), and (4) WBC > 12,000, <4,000 or >10% immature forms (bands) absent any other cause, such as antineoplastic chemotherapy. Sepsis is the characterization of the host response to the insult and thus has a specific definition: SIRS caused by infection. Severe sepsis is sepsis complicated by dysfunction of at least one organ, whereas septic shock is severe sepsis with hypotension (cardiac dysfunction) that is refractory to fluid administration (see Chapter 14). Organ dysfunction is also recognized as a common consequence of SIRS, and the term MODS signifies the presence of altered organ function in critically ill patients such that homeostasis cannot not be achieved without intervention. But which intervention (see below)? Specifically, SIRS correlates with both the incidence and magnitude of MODS, and ultimately mortality.27
Currently, MODS remains the acronym used most commonly to describe organ dysfunction during critical illness. However, several other terms have been used, and may remain in use (Table 48.1).28–31 No scoring system is “right or “wrong”; indeed, scores such as the MOD score28 (Table 48.2) and the Sequential Organ Failure Assessment (SOFA) score30 quantify largely the same pathophysiology and behave similarly.32–35 However, the absence of a validated score that is recognized universally leads to conflicting reports of the incidence, time course, and mortality of MODS, hampering both research and clinical understanding.36 Consensus is needed urgently.
TABLE 48.1
HISTORIC SYNONYMS FOR WHAT IS REFERRED TO CURRENTLY AS THE MODS

MODS, multiple organ dysfunction syndrome.
TABLE 48.2
THE MULTIPLE ORGAN DYSFUNCTION SCORE
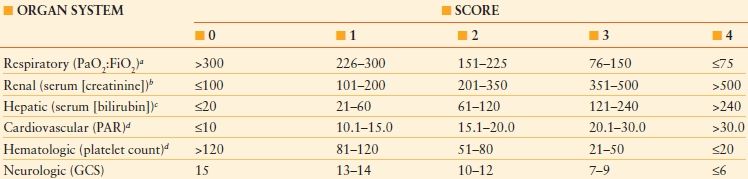
aPaO2:FiO2 is calculated without reference to the use or mode of mechanical ventilation, and without reference to the use or level of positive end-expiratory pressure.
bThe serum creatinine concentration is measured in mmol/L, without reference to the use of dialysis.
cThe serum bilirubin concentration is measured in mmol/L.
dThe platelet count is measured in platelets/mL × 10-3; PAR, pressure adjusted heart rate; GCS, Glasgow Coma Scale score. To convert the serum creatinine concentration from mmol/L to mg/dL, divide by 88.4. To convert the serum bilirubin concentration from mmol/L to mg/dL, divide by 17.1.
Reproduced from Marshall JC, Cook DJ, Christou NV, et al. Multiple Organ Dysfunction Score: a reliable descriptor of a complex clinical outcome. Crit Care Med. 1995;23: 1638–1652.
EPIDEMIOLOGY
The MODS is the leading cause of death among nontrauma ICU patients.37–40. As many as 19% of ICU patients will develop MODS,35,37–40 and MODS is responsible for approximately 50%41,42 to 80%-94%Q338,43,44 of ICU mortality. Patients who develop MODS experience a 25-fold increase in mortality43 and a doubled length of stay compared to critically ill patients who do not develop organ dysfunction.45 MODS is the most common ICU diagnosis in patients associated with prolonged stay (>21 days) in the ICU;46 even modest degrees of MODS47 prolong hospitalization.
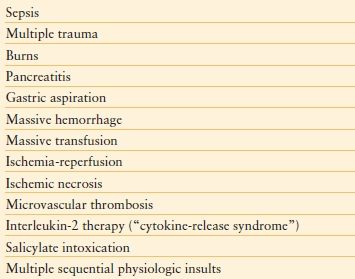
Any biologic stress that activates systemic inflammation may precipitate SIRS, thus placing the patient at risk of MODS. Known precipitants of MODS are listed in Table 48.3; hypoperfusion/I-R injury without shock was the most common etiologic insult responsible for MODS in one study, followed by sepsis without shock, and shock regardless of etiology.45 In another prospective study of elderly patients with ARDS, I-R injury was the leading cause.48 Moreover, Sauaia et al.49 found that an Injury Severity Score (ISS) of ≥ 25 points combined with a transfusion requirement of ≥ 6 units of red blood cell concentrates was associated with a 46% likelihood of developing MODS. Cryer et al.50 further defined these risk factors, reporting a 66% incidence of MODS in patients with an ISS ≥ 25, regardless of transfusion requirement. For any given mechanism, both increased age39 and number of comorbidities39,51 increase the likelihood of MODS. Population data indicate that black persons may have higher case rates of infection, severe sepsis, acute organ dysfunction, and mortality.52 Critically ill children may also develop MODS.53,54 The pathophysiology appears to be similar53 (see below), and neonates and younger children appear to be at higher risk than older children.54
TABLE 48.3
CAUSES OF THE MODS
Furthermore, certain individuals may harbor a genetic predisposition to MODS in the form of an exaggerated innate immunity/inflammatory response to illness.55–58 Single nucleotide polymorphisms (SNPs) have now been identified for a number of genes and peptides related to pattern recognition receptors (PRRs), signal transduction molecules, effector cytokines, and coagulation proteins (Table 48.4), but owing to their multiplicity (with more likely to be identified), it is unlikely that causality will be established for a single SNP or combination. However, their assay does hold promise as a biomarker of risk, predisposition, or the development of MODS.
TABLE 48.4
SNPS IDENTIFIED IN THE PATHOGENESIS OF TRAUMA, SEPSIS, AND MODS
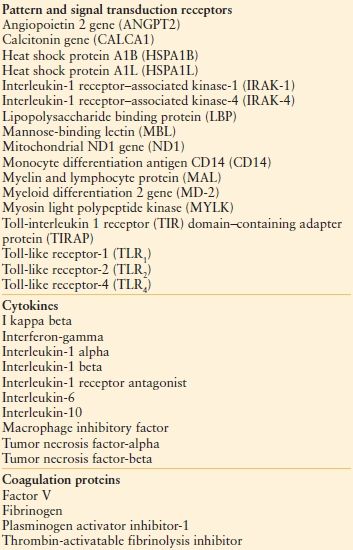
MODS, multiple organ dysfunction syndrome.
PATHOPHYSIOLOGY
Several phenotypes have been proposed to describe the onset of MODS59 (Algorithm 48.1). According to the one-hit model , organ dysfunction develops as the direct result of a massive initial insult (e.g., burn or multiple injuries, severe pancreatitis), which itself is sufficient to cause MODS. By contrast, the two-hit model describes sequential insults, usually isolated temporally. According to the two-hit model, a “priming” insult (e.g., burn) is followed by a subsequent insult (e.g., central line–associated blood stream infection). The inflammatory response, “primed” by the first insult to react in an exaggerated manner to the second, induces further immune dysfunction and MODS. According to the sustained-hit model , a continuous, smoldering insult, such as ventilator-associated pneumonia caused by a difficult-to-treat, multi-drug-resistant bacterium, at once causes and sustains organ dysfunction. In reality, any of these mechanisms, alone or in combination, may result in MODS.
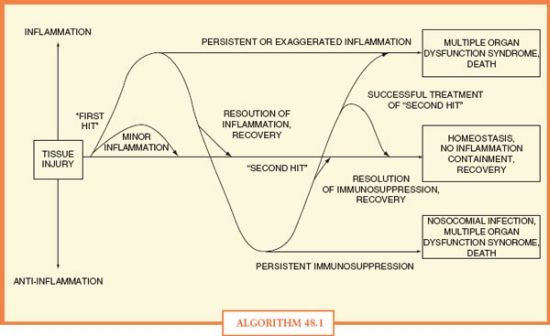
ALGORITHM 48.1 Ebb and flow of pro- and anti-inflammatory responses to tissue injury, resulting in recovery, organ dysfunction, nosocomial infection, or death.
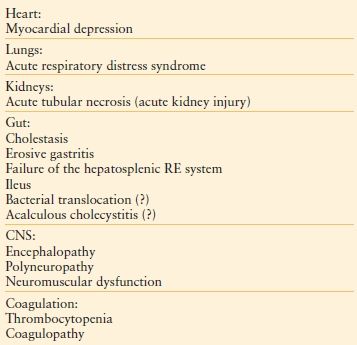
Organ failure may manifest in several organ systems (Table 48.5), and is no longer believed to follow a particular temporal sequence. Cardiovascular instability is often the first manifestation of dysfunctional homeostasis, resulting in I-R injury after resuscitation. The splanchnic and renal circulations are particularly susceptible to I-R injury. Pulmonary failure is equally common and is usually an early manifestation, whereas hepatic, hematologic, and GI dysfunctions and acute kidney injury usually are later manifestations, if they occur at all.9,14,43,60 In particular, hepatic dysfunction may not be recognized promptly because the liver has redundant metabolic capacity; substantial hepatic dysfunction may precede elevation of the serum bilirubin concentration. Furthermore, certain combinations of organ failure have been shown to be especially deleterious (e.g., hepatic and pulmonary, or renal and pulmonary).10,11
TABLE 48.5
EFFECTS OF SHOCK AND SEPSIS ON ORGAN FUNCTION: CLINICAL MANIFESTATIONS
Two distinct periods of altered immune function characterize MODS. The first is dominated by upregulated innate immunity (Table 48.6), uncontrolled inflammation, increased endothelial permeability, microvascular thrombosis, apoptosis (programmed cell death), and disruption of parenchymal cellular integrity.61–63 The second involves a predominance of anti-inflammatory cytokines, downregulated adaptive immunity (Table 48.6), immunosuppression,64 and an increased risk of infection. This general disruption of the normal regulation of the immune system during MODS has been termed immunologic dissonance.65
TABLE 48.6
BASIC FUNCTIONS OF THE INNATE AND ADAPTIVE IMMUNE SYSTEMS

Inflammation and Tissue Injury
Organ damage following severe injury is believed to occur secondary to uncontrolled activation of the inflammatory response caused by tissue hypoxia. Following tissue injury, the early stage of the inflammatory response is characterized by macrophage activation as well as secretion of inflammatory cytotoxins and cytokines. Cytotoxins are released primarily by cluster of differentiation 8 (CD8)+ cells (e.g., cytotoxic T cells, dendritic cells), a cell surface co-receptor for major histocompatibility Class II (MHC Class II); these cells act locally by causing damage to cell walls and tight junctions.66 By contrast, cytokines mediate primarily the CD4+ (a cell surface co-receptor for MHC Class I) response, and may be secreted by a variety of cell types (e.g., platelets, endothelial cells) in addition to those of the immune system (e.g., T helper [Th] cells, regulatory T cells, monocytes, macrophages, and dendritic cells). Cytokines may act both locally and systemically. Although they may be classified broadly into groups by function (Table 48.7), the cytokine-mediated inflammatory response is redundant; each cytokine has multiple activities on different cell types, some of which are salutary. The balance of pro and anti-inflammatory responses is not always self-regulatory and balanced; predominance of either influence may be deleterious. As such, the inflammatory response has been challenging to manipulate for therapeutic effect.
TABLE 48.7
MAJOR CLASSES OF INFLAMMATORY CYTOKINES, REPRESENTATIVE MEMBERS, AND THEIR MAIN ACTION(S)
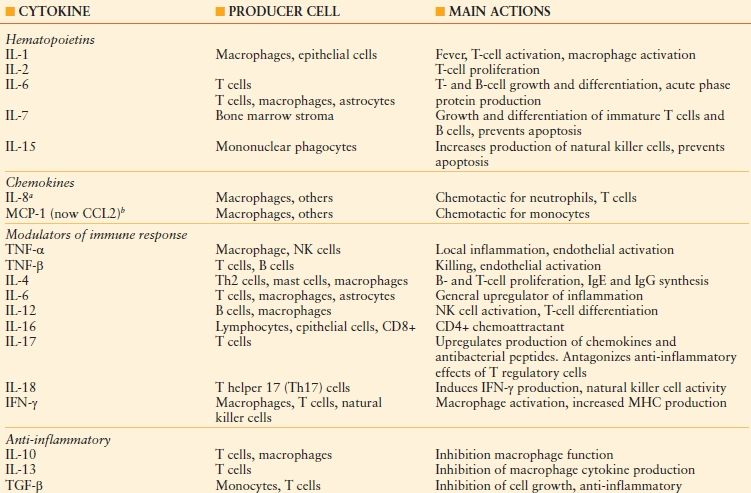
IL, interleukin; MCP, macrophage chemoattractant protein; TNF, tumor necrosis factor; NK, natural killer; IFN, interferon; MHC, major histocompatibility complex; TGF, transforming growth factor.
aSeventeen CXC chemokines (so named because one amino acid separates two cysteine molecules) have been identifi ed in this subgroup of the chemokine family, of which IL-8 is one, along with seven G protein-coupled receptors.
bTwenty-seven CC chemokines (so named because of the adjacency of two cysteine molecules) have been identifi ed in this subgroup of the chemokine family. CCL2: CC ligand 2.
Recruitment of polymorphonuclear (PMN) leukocytes into tissue represents a second prominent feature of MODS. Postmortem examination of patients with ARDS has demonstrated massive PMN leukocyte infiltration into lung parenchyma.67 Neutrophil activation (respiratory burst) generates large amounts of reactive oxygen species and lipid mediators (e.g., prostaglandins, leukotrienes) that are autodestructive. Furthermore, depletion68 or inhibition69,70–71 of PMN leukocytes decreases the severity of lung injury in animal models of ARDS. Infiltration of PMN leukocytes is accompanied by upregulation of hepatic inflammatory proteins (e.g., C-reactive protein) and the complement system,72 increased capillary permeability, and the formation of reactive oxygen and nitrogen species (Table 48.8). The clinical manifestation of increased capillary permeability is tissue edema, whether of connective tissue, lung, or brain.
TABLE 48.8
OXIDANTS AND TISSUE INJURY

Production of oxidizing species plays a central role in killing of pathogens, with activated phagocytes producing both reactive oxygen and nitrogen species. These include superoxide (•O2–), nitric oxide (•NO) and their particularly reactive product, peroxynitrite (ONOO–). This effect is non-specific; almost every part of the target cell is damaged, preventing a pathogen from escaping destruction by mutation of a single molecular target, but damage to host tissues is problematic.
Innate immune cells have evolved to sense microbial pathogens through PRRs, which interact with conserved pathogen-associated molecular patterns (PAMPs) to convey microbial information into immune cell signaling and activation events.73 PRRs also recognize endogenous damage-associated molecular patterns (DAMPs) (also called alarmins or endokines) released during microbial invasion, mediated in part by Toll-like receptors (TLRs). The DAMP molecules, including high mobility group box-1 protein (HMGB1), heat-shock proteins (HSPs), uric acid, altered matrix proteins, and S100 proteins, represent important danger signals that mediate inflammatory responses through the receptor for advanced glycation end-products (RAGE) and TLRs, after release from activated or necrotic cells. The HMGB1 protein is released actively after stimulation of the innate immune system by PAMPs and is released passively by sterile ischemia or cell injury.74–77 The RAGE is a multiligand MHC Class III receptor of the immunoglobulin gene superfamily. Infection is associated with the release of HMGB1 and S100A12. Engagement of RAGE by its diverse ligands, which include HMGB1, results in receptor-dependent signaling and activation of nuclear factor-kappa beta (NF-kB). Furthermore, RAGE acts as an endothelial adhesion receptor for leukocyte integrins and promotes leukocyte recruitment. Inhibition of RAGE signaling reduces inflammatory responses in several noninfectious and infection models of inflammation.
A key question concerns how sterile injury activates innate immunity to mediate damaging inflammation in the absence of invasion by pathogens. That HMGB1, a ubiquitous nuclear protein, mediates the activation of innate immune responses, led to the understanding that HMGB1 plays a crucial role at the intersection of the host inflammatory response to sterile and infectious threat through TLR4 signaling pathways that mediate cytokine release and tissue damage. The RAGE-HMGB1 complex also regulates autophagy (programmed cell survival) and apoptosis (programmed cell death), sustaining the former and inhibiting the latter.
Apoptosis is deranged in MODS. Markers of apoptosis such as the Fas receptor (also known as tumor necrosis factor [TNF] receptor superfamily member 6 and CD 95)78 (a cell-surface protein receptor expressed on essentially all cells of the body that when bound to its ligand [FasL] is internalized and signals an initiator caspase cascade [caspase-8, -10], ultimately resulting in apoptosis), and nuclear matrix proteins79 have been identified in patients with MODS. Interestingly, whereas some cell types (e.g., splenocytes and hepatocytes) demonstrate accelerated apoptosis, other cell types, such as circulating neutrophils, exhibit inhibited apoptosis. Still other tissues, such as the kidney and lung, show only minimal changes in rates of cellular death.80–82
There are two major pathways of apoptosis: The death-receptor pathway , which is mediated by activation of death receptors and the B-cell leukemia/lymphoma 2 (Bcl2)-regulated mitochondrial pathway, which is mediated by stimuli that ultimately lead to mitochondrial injury.83 Ligation of death receptors recruits the adaptor protein, Fas-associated death domain, which in turn recruits caspase-8. This latter ultimately activates caspase-3, the key “executioner” caspase. In the mitochondrial pathway, proapoptotic Bcl homology (BH)-3 proteins are activated by noxious stimuli, which interact with and inhibit antiapoptotic Bcl2 or Bcl-extra large (BCL-XL) proteins. Thus, Bcl2-associated X protein (BAX, a proapoptotic antagonist of Bcl2) and BAK (Bcl-2 homologous antagonist/killer) act unopposed to alter transmembrane potential and induce mitochondrial permeability with release of cytochrome c, which ultimately results in the activation of caspase-9 through the apoptosome. Caspase-9 then activates caspase-3.
The uncontrolled inflammation characteristic of MODS is accompanied by microvascular thrombosis.84,85 Indeed, the inflammatory and coagulation systems are related intimately,85 manifesting as failure of the microcirculation. Several proinflammatory cytokines (e.g., TNF-α) may activate tissue factor and initiate the coagulation cascade.86 In turn, the thrombin receptor activates nuclear factor kappa beta (NF-κB), which causes increased transcription of proinflammatory gene products.87 Microvascular thrombosis results in tissue hypoxia, thus perpetuating the inflammatory response. Early coagulopathy increases the risk of developing MODS.87
Anti-inflammatory Response
Whereas the initial phase of MODS is characterized by a disruption of homeostatic mechanisms to favor inflammation, a second, distinct later period is characterized by impaired adaptive immunity and increased susceptibility to infection.88 This phenomenon has been called the compensatory anti-inflammatory response system, and the result referred to as immunoparalysis.65 During this later period, increased elaboration of anti-inflammatory cytokines such as IL-10 and -13 and transforming growth factor (TGF)-β, impaired antibody synthesis, and anergy of T lymphocytes are characteristic.89–91
In many cases of sepsis, failure of the immune system to eradicate pathogens presages a prolonged phase of immune suppression, characterized not only by failure to eradicate the primary infection but also by development of secondary nosocomial infections.88 The immune suppression is mediated by multiple mechanisms, including massive apoptosis-induced depletion of lymphocytes and dendritic cells, decreased expression of the HLA-DR cell-surface antigen-presenting complex, and increased expression of the negative costimulatory molecules programmed death 1, cytotoxic T-lymphocyte–associated antigen 4, and B-and T-lymphocyte attenuator and their corresponding ligands. Numbers of regulatory T cells and myeloid-derived suppressor cells are increased, and there is a phenotypic shift from inflammatory type 1 helper T (Th1) cells to an anti-inflammatory phenotype of type 2 helper T (Th2) cells that is associated with the production of IL-10. The innate and adaptive immune systems are both compromised severely with poorly functional “exhausted” CD8+ and anergic CD4+ T cells.
An alternative, systems biology-derived theory describes the pathogenesis of MODS as a disruption of interorgan or intercellular communication.92 Accordingly, each organ system is viewed as a stochastic (random) biologic oscillator, whose activity varies periodically with time. The dynamic behavior of any one organ necessarily reflects the state of the organism as a whole. During normal homeostasis, variability within each oscillator is preserved through mechanical, neural, hormonal, and immune (e.g., cytokine and prostaglandin) inputs. As a result of massive physiologic insult, interoscillator communication becomes uncoupled, resulting in a regularization of normally variable organ outputs.
The majority of research in the area of biologic oscillators has been conducted using loss of normal heart rate variability as a marker of uncoupling. Clinical investigations have reported increased cardiac regularity after administration of endotoxin to healthy volunteers,93 as well as in emergency department patients with sepsis.94 Furthermore, low heart rate variability correlates with both ICU mortality95,96 and mortality from MODS.97,98 As a result, measurement of heart rate variability has emerged as a noninvasive, accurate, and validated tool to predict outcomes during critical illness.99







Full access? Get Clinical Tree


