Chapter 13 Multiple organ dysfunction syndrome
The incidence of multiple organ dysfunction syndrome (MODS) is increasing and accounts for up to one-half of deaths in intensive care. Some 50 years ago, multiple organ failure did not exist as a clinical entity. Patients could not be kept alive long enough for the sequential disturbances in the function of distant organs to occur. In the 1960s, acute respiratory failure characterised by bilateral infiltrates on chest radiograph, now termed acute respiratory distress syndrome (ARDS), was described following a variety of non-pulmonary insults. Finally, in 1973, the first description of multiple organ failure appeared in the surgical literature, describing the course of three patients who subsequently died following surgery for ruptured aortic aneurysm.1
Critical illness is often associated with a downward spiral through a systemic inflammatory response syndrome (SIRS) towards frank organ failure and death. Since organ failure is not an all-or-nothing phenomenon, and because dysfunction usually precedes and may progress to gross organ failure, the old term ‘multiple organ failure’ was deemed unsatisfactory. The American College of Chest Physicians/Society of Critical Care Medicine (ACCP/SCCM) consensus conference in 1992 proposed that the term ‘dysfunction’ identifies a phenomenon in which organ function is not capable of maintaining homeostasis (Table 13.1).2 However, it should be remembered that it is only a descriptive definition and does not provide any insight into the aetiology or pathogenesis of MODS. In fact, it is not clear whether MODS is a single pathological process with variable clinical expression, or a limited phenotypic expression of a large number of pathological processes. As yet no therapeutic interventions specifically directed at MODS have significantly altered outcome.
Table 13.1 Definition of systemic inflammatory response syndrome (SIRS) and multiple organ dysfunction syndrome (MODS)3
| Systemic inflammatory response syndrome |
| The systemic inflammatory response to a variety of severe clinical insults is manifest by two or more of the following conditions: |
| Temperature > 38°C or < 36°C |
| Heart rate > 90 beats/min |
| Respiratory rate > 20 breaths/min or PaCO2 < 32 mmHg (or ventilator dependence) |
| White blood cell count > 12 000 cells/mm3, < 4000 cells/mm3 or > 10% band forms |
| Multiple organ dysfunction syndrome |
| The presence of altered function involving at least two or more organ systems in an acutely ill patient such that homeostasis cannot be maintained without intervention |
AETIOLOGY
Uncontrolled infection was classically thought to be the main precedent leading to SIRS and subsequently to MODS. In an Australian epidemiological study, infection was not identified in 912 of a total of 1803 SIRS episodes.4 Therefore it is thought that a substantial proportion of cases of MODS are not initiated by infection. Table 13.2 lists many of the causes of MODS, but is not exhaustive. It must be noted that any number of these causes may in fact act as a secondary insult precipitating MODS. The common features of non-infective aetiologies of MODS are ischaemia, hypoxia, cytokine release, mechanical injury or any combination of these. Thus MODS may occur after all forms of shock and compartment syndromes. Other important causes include trauma, major surgery, burns, pancreatitis, hepatic failure, pulmonary aspiration syndromes and mechanical ventilation.5 Less commonly, cardiopulmonary bypass, blood product and cytokine infusions, and some drug reactions have been associated with MODS.6
Table 13.2 Causes of multiple organ dysfunction syndrome (MODS)
| Infectious agents | Non-infectious insults | Mechanism |
|---|---|---|
| Bacteria | Mechanical ventilation | Ischaemia |
| Viruses | Aspiration | Hypoxia |
| Fungi | Surgery | Cytokine release |
| Protozoa | Burns | |
| Reperfusion syndromes | ||
| Visceral ischaemia | ||
| Pancreatitis | ||
| Hepatic failure | ||
| Cardiopulmonary bypass | ||
| Massive transfusion | ||
| Transfusion reactions | ||
| Hyperthermia | ||
| Malignancy |
PATHOPHYSIOLOGY
INFLAMMATION
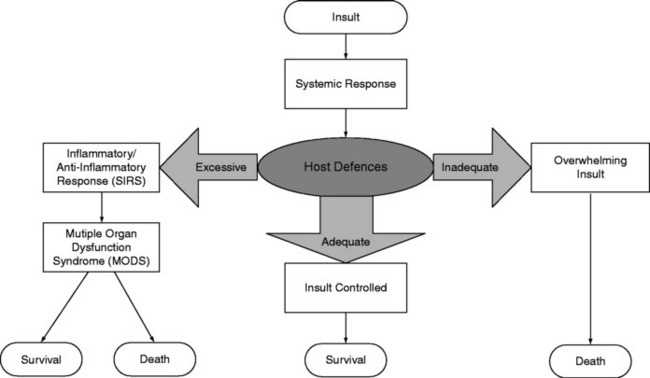
The current explanation for the development of SIRS and MODS is local inflammation with activation of the innate immune system and subsequent unbridled systemic inflammation.7 There is a spectrum of clinical sequelae, ranging from the mildest SIRS that just meets the definition, through organ dysfunction that resolves within a few days, to overwhelming SIRS and MODS. The balance between the inflammatory and regulatory arms of the host response may in part explain these varied responses.
Inflammation involves the activation of circulating immune cells (in particular natural killer (NK), T and B cells and macrophages), the endothelium, and multiple mediator cascades balanced by an anti-inflammatory system. Following injury, proinflammatory mediators are released locally to combat foreign antigens and promote wound healing. Concurrently, anti-inflammatory mediators are released to downregulate this process. If the regulatory mechanisms are unable to contain the response, then inflammatory mediators enter the systemic circulation, additional leukocytes are recruited and activated, and a whole-body response ensues. Homeostasis may be lost and so-called ‘immunological dissonance’ occurs, when the host’s inflammatory or anti-inflammatory response to injury (or both) is excessive or inadequate (Figure 13.1).8 Thus, MODS is not necessarily due to the primary insult, but more likely to be related to an uncontrolled, aggressive systemic response of the host to that primary insult. Without intervention, SIRS may lead to MODS and death.
MOLECULAR MECHANISMS
In sepsis, activated cell/antigen products interact with a member of the Toll-like receptor family to signal gene expression of inflammatory mediators. Toll-like receptors are evolutionarily-conserved receptors expressed by monocytes and macrophages, which recognise whole subsets of pathogens, and represent the phylogenetically ancient innate immune system. Toll-like receptors mediate cell signalling through the same intracellular transcription pathways as cytokines themselves, most notably nuclear factor κB (NF-κB).10 Non-infective causes of MODS seem to have this final common pathway, resulting in mediator release. For example, in ventilator-associated lung injury, the signalling cascade converting the physical stimulus into mediator release operates via NF-κB, but the upstream events remain unclear.11 Large numbers of mediators are involved in inflammation, with chemotactic factors attracting, adhesion molecules focusing, and cytotoxic agents assisting this process. They include cytokines, leukotrienes, prostaglandins, platelet factors and the coagulation and complement systems.
Cytokines are the main mediators of inflammation, with several actions, including directing the cellular response, inducing enzyme production such as inducible nitric oxide synthase (iNOS) and altering adhesion molecules. Their actions are pleiotropic, acting on multiple target cells in different ways depending on timing and local tissue concentration. Several cytokines have been implicated in the development of SIRS and MODS, including tumour necrosis factor (TNF)-α, and interleukins (IL) 1β, 6, and 8. Cytokine and NF-κB concentrations appear to be linked to morbidity and mortality.12,13 In response to proinflammatory mediators, there is endogenous production of anti-inflammatory cytokines such as IL-4, IL-10 and IL-13.
However, it seems that the prevailing internal milieu is likely to be more important than any absolute levels of one mediator or another. Patients at increased risk of SIRS and MODS, such as the elderly and those with pre-existing illnesses, seem to have abnormal cytokine levels.14 The ability of a cell to synthesise pro- or anti-inflammatory mediators is influenced by many factors, both genetic and environmental in origin, but also by its previous state of activation. It may be that an initial insult, insufficient to cause MODS, nevertheless pre-primes a susceptible individual such that a subsequent or secondary insult generates a response that overwhelms homeostasis.
If a relative excess or deficiency of mediator expression can upset inflammatory homeostasis, then it is not surprising to find a strong genetic correlation. Families characterised by low TNF-α production have a 10-fold increased risk of fatal outcome in meningococcal disease, whereas high IL-10 production increases the risk 20-fold.15 TNF-α and IL-10 receptor antagonist polymorphisms are associated with greater susceptibility and worse outcomes to severe sepsis.16 Unfortunately genetic determinants are likely to be more complicated than simple quantitative expression of one mediator or another.
TISSUE INJURY
The final common pathway leading to MODS is often tissue hypoxia. Many factors can compromise oxygen delivery to tissues. Among these factors are arterial hypoxaemia due to acute lung injury, and reduced cardiac output from reduced left ventricular preload and/or impaired ventricular performance. Over and above oxygen delivery abnormalities, evidence is rapidly building that microcirculatory and mitochondrial dysfunction with an abnormal distribution of blood flow and defective oxygen utilisation (‘tissue dysoxia’) is central to the pathogenesis of MODS. Indiscriminate injury by mediators of inflammation leads to deranged endothelial function with altered vascular relaxation and abnormal modulation of coagulation, and mitochondrial and cellular damage. This is compounded by an ongoing initial or subsequent insult. Unchecked, cellular dysfunction leads to loss of ion gradients, leakage of lysosomal enzymes, proteolysis and cell death. If enough tissue injury occurs, organ dysfunction and ultimately failure ensue.
ENDOTHELIAL DYSFUNCTION
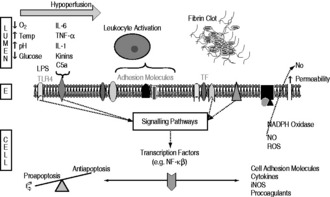
The endothelium is activated by the engagement of antigen, for example lipopolysaccharides from bacterial cell walls, with Toll-like receptors or inflammatory mediators with various receptors. At any stage other environmental factors such as hypoxia, hypoperfusion, increased temperature, acidosis and glycaemic perturbations may also affect endothelial function. The interaction of these extracellular factors with their receptors activates downstream signalling pathways, influencing transcription factors and altering cellular function and/or gene expression. Cell surface adhesion molecules are upregulated, leading to increased leukocyte rolling, adhesion and transmigration, with cytokine-mediated positive feedback and cellular recruitment enhancing this process.17,18 Upregulation of iNOS leads to excessive amounts of NO production.
In addition, inappropriate intravascular coagulation is an important cause of tissue injury. Activation and amplification of the tissue factor pathway, and possibly downregulation of anticoagulatory pathways, lead to the generation of thrombin and hence fibrin formation. Coagulation may result in focal areas of ischaemia, and concurrent depletion of counterregulatory networks. The mechanisms that regulate inflammation, coagulation and the various cell types are intimately linked and cannot be thought of as discrete entities. A schematic diagram of the processes involved in endothelial dysfunction is shown in Figure 13.2.
Relatively new non-invasive techniques such as orthogonal polarisation (Figure 13.3) have demonstrated these abnormalities in microcirculatory blood flow distribution in septic states compared with normal controls. As a consequence of the derecruitment of capillaries, the increased diffusion distances will in turn contribute to the development of hypoxia.
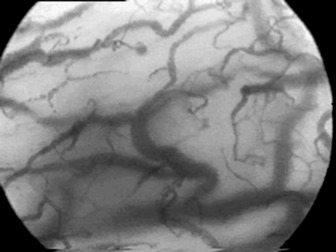
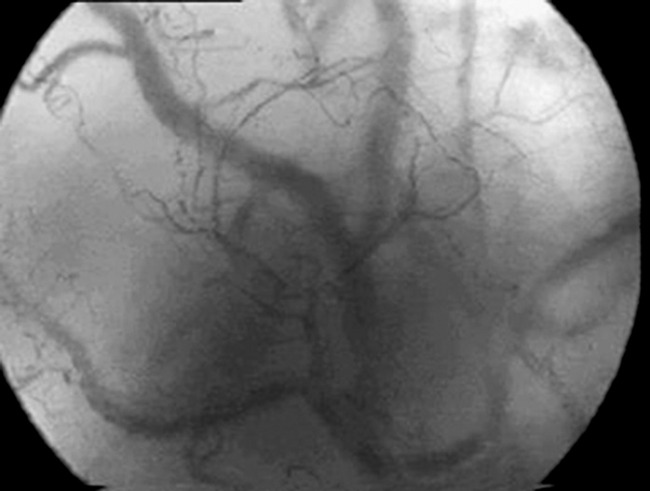
Figure 13.3 Orthogonal polarisation images of the sublingual mucosa in a normal (top) and a septic (bottom) patient. Note, firstly, paucity of smaller blood vessels in the septic sublingual mucosa with increased diffusion distances between blood vessels and, secondly, that the remaining blood vessels are of smaller calibre.
(Courtesy of Cytometrics.)





Full access? Get Clinical Tree


