Chapter 117 Molecular Mechanisms of Drug Actions
From Receptors to Effectors
Optimizing drug response is a challenging task that clinicians confront on a daily basis. This is particularly true for those caring for critically ill patients, in whom many factors influencing drug response are being more commonly recognized. These include reduced absorption, variable drug distribution, decreased metabolism and elimination, as well as alterations in drug receptors, signaling mechanisms, and effectors.1–3 In recent years, advances in molecular pharmacology have shed more light on the processes that transduce extracellular signals into intracellular messages that control cell function. This has led to the elucidation of multiple points at which modulation of signal transduction, either by pharmacologic agents or diseases, can occur. Also, there has been an ongoing recognition of the role of inheritance in the individual variation of drug response with the identification of polymorphisms in genes encoding drug-metabolizing enzymes, drug targets (e.g., receptors, enzymes), and proteins involved in signal transduction.4–6
This chapter provides a detailed overview of how drugs work at the molecular level and how this complex system is influenced by genetic factors, developmental changes, disease processes, and the environment (Figure 117-1). Ultimately, the objective is to help pediatric intensive care physicians better tailor the pharmacotherapy they use: that is, choose the right drug, or combination of drugs, for the right patient to achieve maximal efficacy with no or minimal toxicity. This chapter does not address signaling pathways involved in diseases per se.
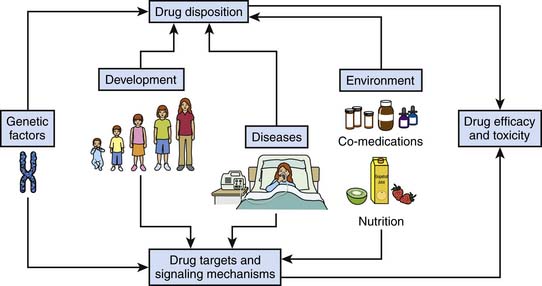
Figure 117–1 Relationships among genetic factors, development, diseases, and the environment and drug efficacy and toxicity.
Targets For Drug Action
The initial step in the cascade of biochemical events resulting in drug action mostly consists in the binding of drugs to specific cellular targets. These can be broadly divided into four categories: (1) receptors, (2) ion channels, (3) enzymes, and (4) carrier proteins (Figure 117-2). The vast majority of important drugs act on one of these types of proteins.
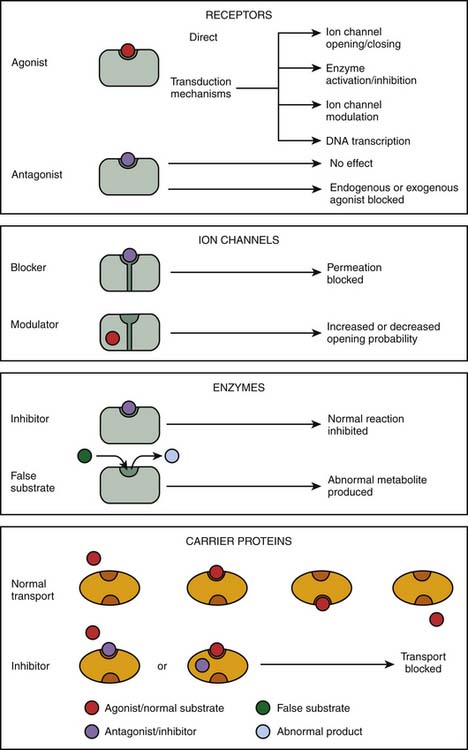
Figure 117–2 Targets for drug action.
(Modified from Rang HP, Dale MM, Ritter JM, et al: Rang and Dale’s pharmacology, ed 6, Philadelphia, 2007, Churchill Livingstone.)
Receptors
Receptors are the most frequent drug target. They can be defined as the sensing elements in the system of chemical communication that coordinate the function of all different cells in the body, the chemical messengers being the various hormones, neurotransmitters, other mediators, or drugs. Ligands (e.g., hormones, drugs) that bind with receptors are termed agonists if their binding results in the expected effect and are termed antagonists if binding stops or decreases an agonist-induced activity.7 Administration of a receptor antagonist in the absence of agonist results in no effect because antagonists bind to receptors but do not activate them. An antagonist is said to be surmountable when maximal response to the agonist can be restored by raising the agonist concentration (parallel displacement of the agonist dose-response curve to the right). Likewise, an antagonist is said to be insurmountable when even high concentrations of the agonist are not able to elicit the maximal expected agonist response (Figure 117-3, A). The terms competitive and noncompetitive do not describe a pharmacologic behavior per se; rather, they refer to the receptor binding site of the antagonist with regard to that of the agonist. A competitive antagonist binds to the same site as the agonist on the receptor, whereas a noncompetitive antagonist has its own binding site separate from that of the agonist and makes the receptor refractory to the agonist. Both competitive and noncompetitive antagonists can be insurmountable.
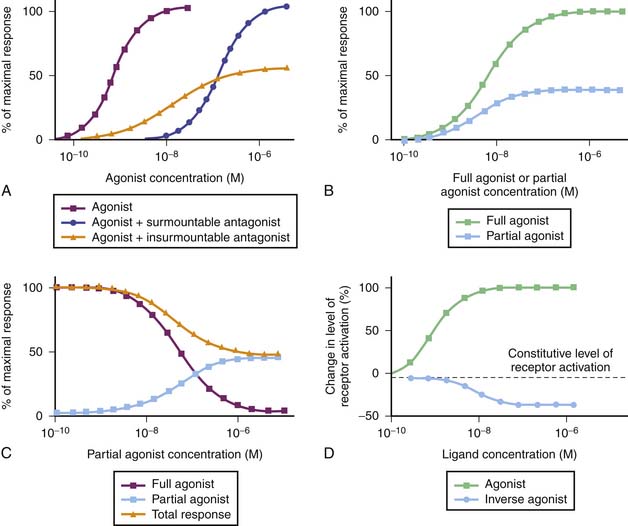
The duration of action of an insurmountable antagonist depends mostly on synthesis of new receptors, which can take several days. This may have clinically important consequences. For example, phenoxybenzamine, an insurmountable α-adrenergic receptor antagonist sometimes used in stage I Norwood procedures to balance the pulmonary and systemic circulations, can produce symptomatic hypotension in some patients. The attenuation or reversal of the decrease in systemic vascular resistance it produces may not be achieved with an α-adrenergic receptor agonist such as dopamine (high dose) or norepinephrine, depending on the dose of phenoxybenzamine given. In such circumstances, the use of a pressor agent that does not act through the α-adrenergic receptor such as vasopressin, which binds on V1 receptors of smooth muscle cells, must be considered.8
Partial agonists are ligands that bind to the same receptor as full agonists but have less intrinsic capacity to produce a response as strong as full agonists, despite full receptor occupancy (Figure 117-3, B). The exact mechanism that accounts for the blunted maximal response seen with partial agonists is unknown. Simultaneous administration of a partial agonist and a full agonist prevents the maximal response usually observed with the full agonist alone because partial agonists have the ability to occupy the receptor population (Figure 117-3, C). Finally, inverse agonists are ligands that reduce the level of constitutive activation encountered in some receptor systems (Figure 117-3, D).9
Enzymes
Enzymes are a specialized class of proteins responsible for catalyzing chemical reactions within the cell and thus are ideal drug targets. Most drugs that alter enzymes activity are substrate analogues of enzymes that inhibit their activity either reversibly (e.g., angiotensin-converting enzyme inhibitors acting on peptidyl dipeptidase) or irreversibly (e.g., acetylsalicylic acid acting on cyclooxygenase). Drugs may also prevent the normal functioning of enzymes. Fluorouracil, an anticancer drug, is a good example of such drug; it is converted into an abnormal nucleotide that inhibits thymidylate synthetase, thus blocking DNA synthesis.
Carrier Proteins
Several biologic elements, such as ions and small organic molecules, are not lipid soluble enough to cross the plasma membrane and require a carrier protein to be transported. In most cases, the transport of organic molecules is coupled to the transport of ions (usually Na+), either in the same direction (symport) or in the opposite direction (antiport). The carrier proteins embody a recognition site that makes them specific for a particular permeating species; these recognition sites can also be targets for drugs whose effect is to block the transport system.25 Indeed, some drugs, such as cardiac glycosides (e.g., digoxin), bind to these carrier proteins and interfere with the transport system. Digoxin is a typical example of drugs that produce their effect by inhibition of an ion pump: blockade of the Na+/K+ ATPase pump.
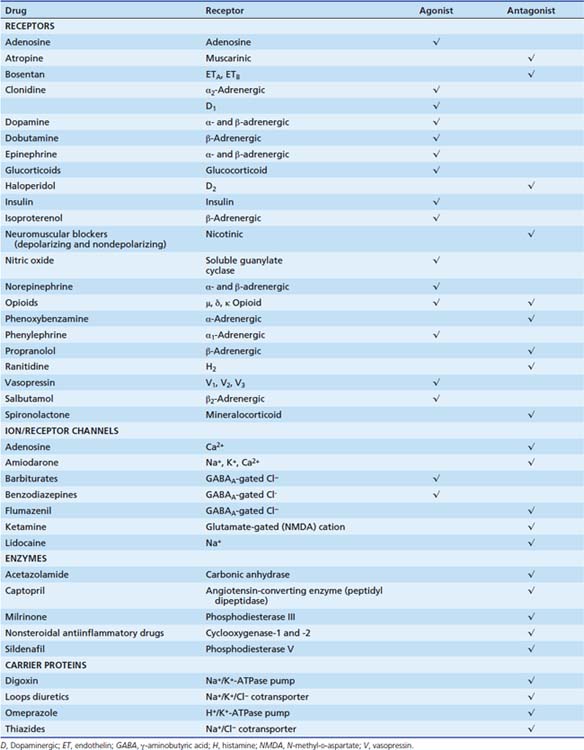
Table 117-1 shows the targets of some pharmacologic agents commonly used in the pediatric intensive care unit.
Receptor Type and Regulation
Classification of Receptors
Four families of receptors, three cell surface receptor types and one nuclear receptor, have been described (Figure 117-4).
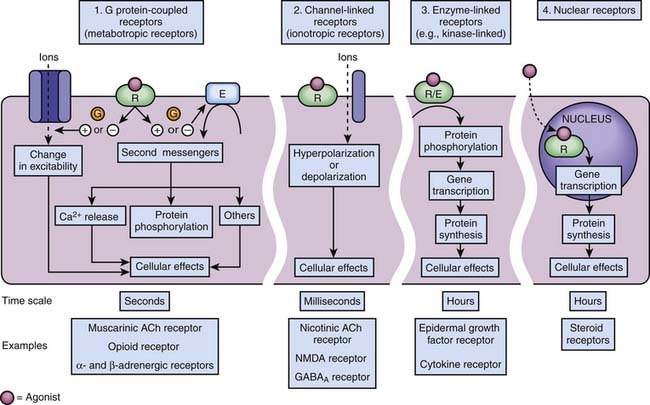
G Protein–Coupled Receptors
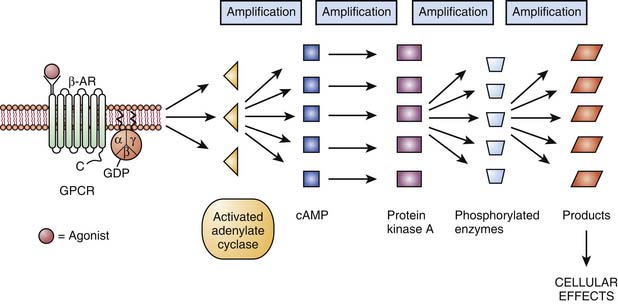
In 1994, Alfred G. Gilman and Martin Rodbell were awarded the Nobel Prize in Physiology or Medicine for their discovery of G proteins and their role in signal transduction in cells.10 G proteins are a superfamily of propeller proteins that allow the transduction between the activated receptor (by an agonist) and different intracellular effectors such as enzymes or ion channels, relaying signals from more than 1000 receptors.11 G protein–coupled receptors (GPCRs), also known as metabotropic receptors, are in fact the first component of the cellular amplification cascade (Figure 117-5). Indeed, the activation of target enzymes through GPCRs leads to the synthesis of numerous second messengers, which in turn activate other enzymes. The intervention of the second messenger system allows for the diversity of the cellular targets (see below). GPCRs represent the target, directly or indirectly, of approximately 50% of all current therapeutic agents.12
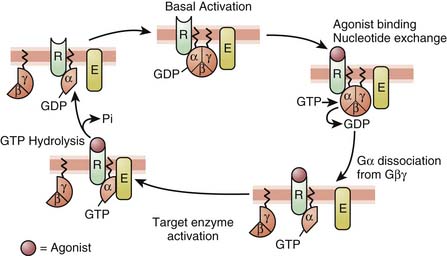
Three major families of GPCRs are defined based on their amino acid sequence: family 1, the largest one, includes receptors for rhodopsin, monoamines (such as β-adrenergic receptors), neuropeptides, opioids, and chemokines. Family 2 consists mainly of receptors for peptides with a large molecular weight, such as calcitonin and secretin. Family 3 has, among others, receptors for glutamate (metabotropic), γ-aminobutyric acidB (GABAB), and extracellular calcium. Despite these differences, the families of GPCRs share characteristic structural and functional features. All GPCRs share a common serpentine structure consisting of seven transmembrane domains with three extracellular and three intracellular loops. The extracellular regions are involved in ligand binding, and the intracellular regions are primarily involved in signaling.13 The latter are coupled to a heterotrimeric guanine-nucleotide-binding regulatory protein (G protein) located on the cytoplasmic portion of the cell membrane and are made of three subunits. Each G protein is composed of an α-subunit that is loosely bound to a tightly associated dimer made up of β- and γ-subunits. The activity of a trimeric G protein is regulated by the binding and hydrolysis of guanosine triphosphate (GTP) by the α-subunit (Figure 117-6).
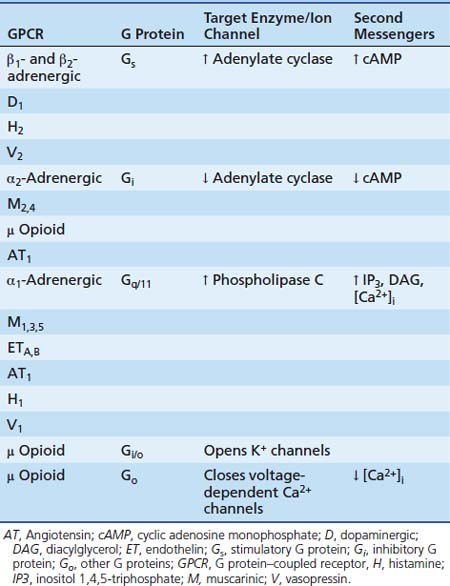
Each of the three subunits is encoded by a separate gene selected from at least 20 α, six β, and 12 γ genes, respectively. The α-subunit is essential in the “receptor-effector” coupling. Various α-subunits define different G protein trimers (Gs = stimulatory G protein, Gi = inhibitory G protein, Go = other G protein, etc.), each of which regulates a distinctive set of downstream signaling pathways. Table 117-2 shows some examples of GPCRs with their trimeric G protein along with their target enzymes or ion channels and second messengers. Some receptors act by way of more than one type of G protein trimer (e.g., μ opioid receptor). Approximately 50% of the GPCRs couple to Gi/Go proteins, approximately 25% couple to Gs, and about the same amount couple to Gq proteins.14 Gs proteins (made of αs-subunits) can activate adenylate cyclase and are inhibited by the cholera toxin. In contrast, Gi proteins (made of αi-subunits) can inhibit adenylate cyclase and open K+ channels and are inhibited by the pertussis toxin.
Table 117–2 Examples of GPCRs with their Trimeric G Protein, Target Enzyme and/or Ion Channel, and Second Messengers
Small G proteins are monomeric G proteins with molecular weight of 20 to 40 kDa. As with heterotrimeric G proteins, their activity depends on the binding of GTP. More than 100 small G proteins have been identified. They are classified into different families: Ras, Rho, Rab, Rap, Ran, and ARF. They play key roles in numerous cellular functions such as cell division, proliferation, differentiation, vesicle trafficking, cytoskeletal reorganization, and gene expression.15
Channel-Linked Receptors
Receptors of excitatory mediators (glutamate, aspartate, and acetylcholine), which comprise the N-methyl-D-aspartate [NMDA] (Figure 117-7, A) and the nicotinic acetylcholine receptors, are receptors whose activation provokes depolarization of the cell, leading to propagation of the action potential and ultimately secretion of a neuromediator and muscular contraction, for example. These receptors are permeable to monovalent and divalent cations, mainly Na+, K+, Ca2+, and magnesium (Mg2+). Ketamine, a dissociative anesthetic, is a noncompetitive surmountable NMDA receptor antagonist that acts by preventing the opening of ion channels by glutamate. In addition, the potential neuroprotective effects of ketamine appears to be mediated via NMDA receptor blockade.
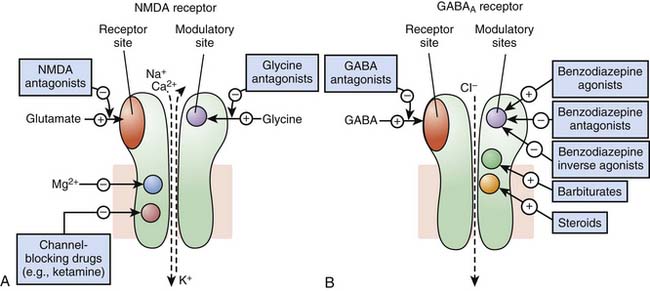
Receptors of inhibitory mediators, the activation of which provokes hyperpolarization of the cell and therefore decreases cellular excitability, are a group that includes GABAA (Figure 117-7, B) and glycine receptors. These ligand-gated ion channels are selective for anions such as chloride (Cl−) or phosphorus (PO43−). GABA is the major inhibitory neurotransmitter in the central nervous system, and drugs that potentiate GABAergic inhibition in the brain, such as benzodiazepines and barbiturates, result in sedation and hypnosis.7 Benzodiazepine agonists enhance Cl− ion conductance induced by GABA by increasing the frequency of channel-opening events, whereas barbiturates seem to do so by increasing the duration of the GABA-gated channel openings. Both classes of agents bind to sites on the GABAA molecule that are different from each other and also from the GABA receptor site.
Enzyme-Linked Receptors
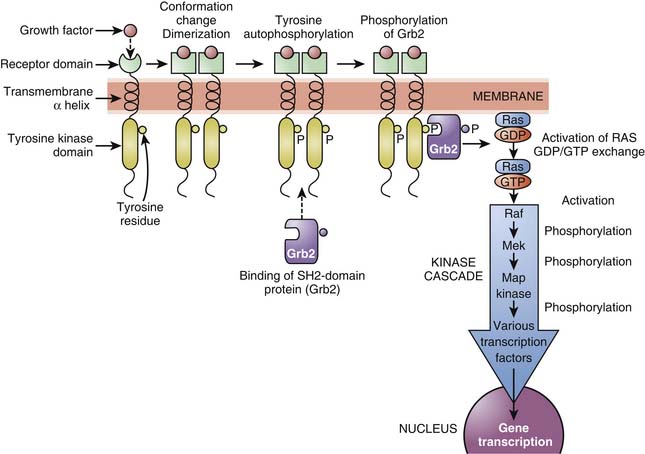
Tyrosine kinase receptors include receptors for neurotrophin,16 growth factors (epidermal growth factor [EGF], platelet-derived growth factor [PDGF]), as well as insulin and many other trophic hormones. These receptors shift from an inactive monomeric state to an active dimeric state upon agonist binding (dimerization). This is followed by autophosphorylation of the intracellular domain of each receptor and binding of SH2-domain proteins that are themselves phosphorylated. Depending on the receptor subtype, SH2-domain proteins allow the phosphorylated receptor to activate other functional proteins, which eventually results in stimulation of gene transcription, or are enzymes such as phospholipases, leading to the formation of second messengers (see below). One important pathway involved in the transduction mechanisms of tyrosine kinase receptors include the Ras/Raf/MAP) kinase pathway which is important in cell division, growth, and differentiation (Figure 117-8).
< div class='tao-gold-member'>






Full access? Get Clinical Tree


