Chapter 83
Metabolic Acidoses and Alkaloses
Physiology and Pathophysiology
Acid-Base Physiology
Metabolism of food ultimately produces acid (H+) and base (HCO3–). A typical American diet, heavy in protein, produces slightly more acid than base, equal to approximately 1 mmol H+ per kg body weight per day; in contrast, a strict vegetarian diet produces a net base. Basic organic anions—for example, citrate and acetate—should be considered equivalent to HCO3– because they produce HCO3– when oxidized. Complete oxidation of carbon-containing compounds yields the volatile acid, carbonic acid (H2CO3), as shown by Equation 1:
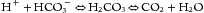
The effect of these metabolic events on acid-base homeostasis is influenced by respiration and the exhalation of CO2. Nonvolatile organic acids (e.g., lactic, beta-hydroxybutyric, and acetoacetic acids) are acids produced by incomplete oxidation and do not yield carbon dioxide. The H+ from nonvolatile acids (e.g., sulfuric and lactic acids) must be excreted by the kidneys, as CO2 is not produced in these reactions.
Buffers minimize pH changes that result from the addition or removal of H+. Equation 1 can be rewritten in the Henderson-Hasselbalch format,
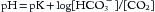
where pK = 6.1 (equilibrium constant for carbonic acid), [HCO3–] = concentration (mmol/L) of bicarbonate ion, and [CO2] is the concentration (mmol/L) of CO2 dissolved in blood. [CO2] is determined by measuring Paco2 and multiplying it by the solubility coefficient (0.03 mm Hg/mmol/L). Thus, a normal Paco2 of 40 mm Hg is equal to 1.2 mmol/L of CO2. When Paco2 is 40 mm and [HCO3–] is also normal (i.e., 24 mmol/L), the ratio of [HCO3–]/[CO2] equals 20:1 (and log 20 = 1.3). This ratio corresponds to the normal pH of 6.1 + 1.3 or 7.40. Remember that it is the ratio of bicarbonate to carbon dioxide in the blood that determines pH—not the absolute value of either.
The [HCO3–]/[CO2] buffer pair is the most important because of the abundance of each part’s components and how the relationship between them can be influenced independently by the kidneys and lungs. For example, by Equation 1, in metabolic acidosis, the addition of H+ increases the concentration of hydrogen ion, [H+] (which, at equilibrium, proportionately decreases the concentration of bicarbonate ion [HCO3–]). By the law of mass action, the increased [H+] drives the reaction to the right, increasing the production of CO2.
Renal Acid-Base Handling
Normally functioning kidneys regulate the serum HCO3– concentration in two ways. First, they reabsorb the filtered load of HCO3– (= GFR × serum [HCO3–]). Losing 1 mmol of HCO3– in the urine would be equivalent to gaining 1 mmol of H+. Second, the kidneys excrete H+ at a rate equal to the metabolic production of H+ and restore the HCO3– consumed in buffering. HCO3– reabsorption occurs in the proximal tubule by a mechanism that depends on carbonic anhydrase to accelerate the hydration reaction of CO2 (Equation 1, with the reaction arrows going right to left). The diuretic acetazolamide inhibits carbonic anhydrase, to increase bicarbonaturia (renal “dumping” of bicarbonate) and often results in metabolic acidosis.
Compensatory Mechanisms
After the onset of a primary acid-base disturbance, compensation mechanisms return blood pH toward normal values. Under most circumstances, primary metabolic abnormalities induce changes in ventilation, and renal acid-base handling changes in response to primary respiratory abnormalities. Although a compensatory process tends to normalize blood pH, it rarely, if ever, returns pH to 7.40. Therefore, the serum pH ultimately reflects which process is the primary disturbance and which is simply compensatory (see the acid-base nomogram [Figure D1] in Appendix D).
Respiratory Compensation for Metabolic Disorders
The respiratory response to acute metabolic acid-base disturbances begins immediately. Hyperventilation compensates for metabolic acidosis, typically characterized by increasing tidal volume, although tachypnea can occur. This pattern can be subtle, but in its most pronounced form it is referred to as Kussmaul breathing/respiration. The magnitude of the compensation varies with the degree of acidosis but can be accurately predicted by “Winter’s formula” for acute metabolic acidosis,
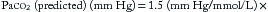
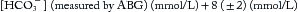
where Paco2 (predicted) is the Paco2 level expected only from respiratory compensation, and [HCO3–] (measured) is the value obtained from an arterial blood gas (ABG) (in which the [HCO3–] is calculated by Equation 2 in Chapter 84 (the Henderson-Hasselbalch equation) from the measured parameters of pH and Paco2)
Hypoventilation with elevation of Pco2 compensates for metabolic alkalosis, proportionate to the alkalosis. However, the hypoventilatory response is more variable than the hyperventilatory response and may be limited by hypoxemia as ventilation slows. Consequently, the compensatory Paco2 is often difficult to predict accurately. The acid-base map or nomogram in Appendix D limits compensatory elevation of Paco2 to 55 mm Hg.
Metabolic Acidosis
The Anion Gap and Elevated Gap Acidoses
The anion gap (expressed in mEq/L) is the difference between measured serum cations and anions, as in Equation 4,
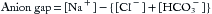
where [HCO3−] is the (total) serum bicarbonate (= dissolved CO2 and HCO3−) as measured on a venous blood sample as part of the same chemistry panel giving values (mEq/L) for serum [Na+] and [Cl–]. The anion gap consists of net negatively charged proteins (albumin is the major contributor) and small anions like urate and phosphate. The normal anion gap ranges from 8 to 12 and increases when nonvolatile acids accumulate because of increased net production (e.g., lactate, acetoacetate, or beta-hydroxybutyrate) or decreased renal excretion (e.g., phosphate and sulfate in chronic renal failure).
Hypoalbuminemia decreases the anion gap. In general, a decrease in albumin concentration of 1 mg/dL will decrease the normal anion gap by approximately 2.5 mEq/L. Paraproteinemias can increase or decrease the gap depending on the charge of the paraprotein. By increasing the net negative charge of albumin, alkalemia widens the gap; acidemia has the opposite effect, and as a result, the anion gap is an underestimate in significant acidemia. Renal disease causes both an anion gap and a normal gap acidosis (Tables 83.1 and 83.2).
TABLE 83.1
Metabolic Acidoses: Anion Gap Acidoses
| Type of Anion | Etiology |
| Ketones | Diabetic ketoacidosis |
| Alcoholic ketoacidosis | |
| Toxins, poisons | Salicylates |
| Methanol | |
| Ethylene glycol Acetaminophen | |
| Lactate | Lactic acidosis |
| Phosphates and sulfate | Chronic renal failure |
TABLE 83.2
Metabolic Acidoses: Hyperchloremic (Nonanion Gap) Acidoses
| Mechanism of Disorder | Etiology |
| Addition of equimolar H+ and Cl– | Ingestion or administration of HCl, NH4Cl, lysine, or arginine HCl |
| Loss of HCO3– with equimolar gain of Cl– | Secretory diarrhea (cholera-like) caused by infections or laxatives; other gastrointestinal loss of base (pancreatic fistula) |
| Inability to excrete daily load of H+ | Renal tubular acidosis (see Table 83.3) |
| Chronic renal failure | |
| Inability to maintain serum [HCO3–] because of renal losses of HCO3– | Renal tubular acidosis (see Table 83.3) Acetazolamide use |
| Dilution of serum [HCO3–] | Volume expansion with fluid loading with NaCl |
Lactic acidosis and diabetic ketoacidosis (DKA) commonly cause an anion gap acidosis in ICU patients (see Table 83.1). In DKA, metabolism of fatty acids produces H+ and the anion acetoacetate. Starvation produces ketosis but only minimal acidosis. In contrast, alcoholic ketoacidosis (AKA), which includes aspects of starvation, can produce a severe anion gap acidosis. Binge drinking leads to an altered redox state that generates the anion beta-hydroxybutyrate (which is not detected by the nitroprusside reaction for ketones, unlike acetoacetate).
Several other toxins induce anion gap acidoses. Salicylates induce lactic acid production while also stimulating respiratory centers to produce a second primary disorder, a respiratory alkalosis. Methanol and ethylene glycol are metabolized by hepatic alcohol dehydrogenase to various toxic metabolites, some of which also elevate the anion gap. Particularly, metabolism of methanol produces the toxins formaldehyde and formic acid. Metabolism of ethylene glycol produces glycolic and oxalic acids. In acute acetaminophen toxicity, glutathione depletion can result in elevated concentrations of gamma-glutamyl cysteine, leading to accumulation of pyroglutamic acid. Suspect acetaminophen toxicity in patients with a large anion gap, hepatic or renal dysfunction, and a negative preliminary toxicologic screen for methanol or ethylene glycol. Inhalation of toluene, a component of paint thinners and many glues, produces an anion gap acidosis via metabolism to hippuric acid. Renal failure leads to an accumulation of phosphate and sulfate, elevating the anion gap, most often late in the disease.


Full access? Get Clinical Tree


