and penicillamine are both used in lead poisoning; penicillamine is also of use in copper and mercury poisoning.
Cyclodextrins are bucket-shaped oligosaccharides produced from starch. α-Cyclodextrins are formed of six, β-cyclodextrins of seven and γ-cyclodextrins of eight sugar residues. They present a hydrophilic outer surface and an internal hydrophobic cavity that can trap other molecules by forming an inclusion complex. Modified cyclodextrins are used extensively for masking odours and as drug delivery systems for poorly water-soluble drugs. Sugammadex is a γ-cyclodextrin reversal agent that selectively forms an inclusion complex with rocuronium and less so with vecuronium but not with benzylisoquinolinium muscle relaxants. This produces effective reversal of rocuronium effects from any depth of neuromuscular blockade without the unwanted effects of widespread inhibition of acetylcholinesterase.
Pharmacodynamic mechanisms
Many drugs exert their effects through an interaction with specific and selective sites on receptors. Receptors are large proteins that are associated with cellular structures such as cell membranes, cytoplasm, intracellular membranes or nuclear material. The selectivity arises from the 3D chemical configuration of the drug, which matches a site on the relevant protein and allows binding to take place. The observed effect then results directly or indirectly from this interaction. This type of action is characterised by a lock-and-key mechanism involving different chemical forces that allow the drug first to approach its active site and then to fit into a selective binding area. Initial attraction may be by ionic forces, but stabilisation is due to van der Waals interactions once the drug is in close proximity to its selective binding site.
Interaction mechanisms include:
Drug–cell membrane receptor
Drug–voltage-gated ion channel
Drug–intracellular membrane receptor
Drug–cytosolic receptor mechanisms
Drug–cell membrane receptors
Many of the drugs we use in our anaesthetic practice act by interfering with the action of endogenous neurotransmitters at their receptors. The most important neurotransmitter-associated receptors are ligand-gated ion channels and G-protein-coupled receptors. Other membrane-dependent transduction mechanisms include tyrosine kinase and guanylyl cyclase-coupled receptors.
Ligand-gated ion channels
Ion channels that are opened as a result of binding of neurotransmitters are known as ligand-gated ion channels. They must be distinguished from ion channels that are opened as a result of a change in membrane potential (voltage-gated channels).
Many of the drugs we use interfere with ligand-gated ion channels that mediate very rapid transmission of information through the central and peripheral nervous systems. Neurotransmitters either depolarise the postsynaptic membrane, allowing forward transmission of electrical signals, or hyperpolarise the membrane, inhibiting such signals.
There are three distinct families of ligand-gated receptors, which can be distinguished by their subunit structure: pentameric, ionotropic glutamate and ionotropic purinergic receptors (Figure 28.1).
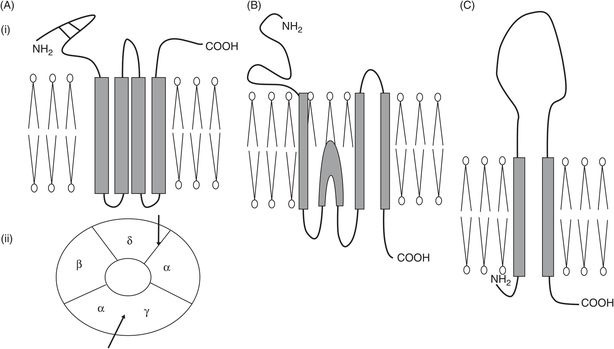
Schematic illustration of the ionotropic receptor families. (A) Pentameric family, typified by nicotinic acetylcholine receptor at the neuromuscular junction; (i) subunit configuration – note four transmembrane domains and two cysteine bridges near the NH2 terminus; (ii) arrangement of subunits as seen from above – arrows show the two acetylcholine binding sites. (B) Ionotropic glutamate family, typified by NMDA receptor with three transmembrane domains and one re-entrant loop. (C) Purinergic ionotropic P2X receptors, with two transmembrane domains and a large extracellular loop.
Cys-loop pentameric receptors
Examples – the nicotinic acetylcholine receptor (nAChR), the γ-aminobutyric acid type A receptor (GABAA), inhibitory glycine receptors (GlyR) and the 5-hydroxytryptamine (serotonin) type 3 receptor (5-HT3).
Each subunit of this pentameric family has four helical transmembrane domains (TMDs). The name cys-loop comes from the fact that in the extracellular N-terminal region there are two disulphide cysteine bridges, forming a looped structure.
The pentameric family provides the most important sites of drug action for neuromuscular blocking agents, at the nAChR, and for many of our general anaesthetic agents, at the GABAA receptor; the action of general anaesthetic agents is discussed in greater detail below.
Subunit composition can vary for the nAChR. At the neuromuscular junction (NMJ), subunit composition is αεαβδ, but in the fetus it is αγαβδ; acetylcholine binds to the α–ε and α–δ subunit interfaces. Cooperative binding of two molecules of acetylcholine is required to produce the required conformational change and open the channel, which is then five times more selective for monovalent cations – Na+ in particular – than for divalent cations such as Ca2+. Depolarising blockers, such as suxamethonium, bind to the same site as the natural transmitter acetylcholine and cause opening of the ion channel by inducing similar conformational changes, although channel opening time is increased. Suxamethonium persists longer in the synaptic cleft than acetylcholine because it is not a substrate for acetylcholinesterase. As a result the receptor cannot return to its resting conformation but becomes desensitised: it no longer responds to the agonist, and neuromuscular blockade ensues. A similar picture is seen in the presence of irreversible inhibition of acetylcholinesterase by organophosphates: acetylcholine itself induces paralysis. In contrast, the non-depolarising muscle relaxants compete for the same binding site as acetylcholine but the conformational change they induce prevents channel opening.
Nicotinic receptors are found at sites other than the NMJ, in particular at autonomic ganglia and in the CNS. In the CNS the subunit composition is very different from that at the NMJ, which accounts for the differing sensitivity of these receptors to cholinergic drugs. As discussed below, these neuronal nicotinic receptors are sensitive to the effects of certain general anaesthetic agents.
GABA and glycine are the major inhibitory neurotransmitters in the CNS, glycine predominantly in spinal cord and hindbrain, GABA supraspinally. Unlike the nAChR, the GABAA channel is an anionic channel favouring chloride passage through the synaptic membrane, resulting in hyperpolarisation and inhibition of forward signalling. Subunit stoichiometry depends on anatomical location, but 1α:2β:2γ and 2α:2β:1γ are most common. The benzodiazepine receptor (BDZR) site is associated with the GABAA–chloride channel complex. Activation of the BDZR is responsible for sedative and anticonvulsant effects due to positive allosteric modulation of GABA transmission, so increasing hyperpolarisation. The benzodiazepine binding site requires both α and γ subunits to be present, whereas etomidate binds with higher affinity to receptors with a β2 or β3 subunit. Important sites for drug binding on the GABAA receptor complex are shown in Figure 28.2.
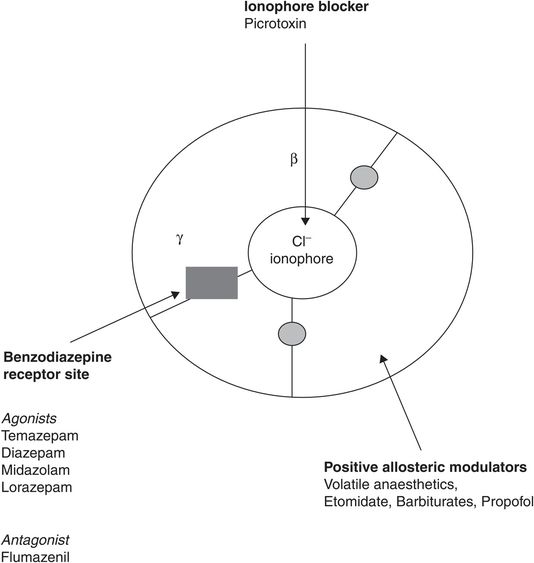
Active sites on the GABAA/Cl– ionophore/benzodiazepine receptor complex, looking from above. The grey circles show the two agonist sites for GABA, which are competitively inhibited by gabazine and bicuculline.
Ondansetron inhibits 5-hydroxytryptamine type 3 (5-HT3) ionotropic channels. Like nAChR, 5-HT3 receptors are cation channels favouring monovalent over divalent cations. There are several types of serotoninergic receptors, but only type 3 are ionotropic; the others are all G-protein-coupled receptors. The centrally mediated antiemetic action is associated with vagolytic effects, as these receptors are also found on vagal afferents from the gastrointestinal (GI) tract. Ondansetron therefore has both central and peripheral effects.
Ionotropic glutamate receptors
There are three ionotropic glutamate receptor types, NMDA, AMPA and kainite. Other glutamate receptors are metabotropic G-protein-coupled receptors. NMDA receptors are formed from four subunits, two are pore-forming (NR1) and two regulatory (NR2). Glutamate binds to the NR1 subunit and its coactivator, glycine, to the regulatory subunit. All ionotropic glutamate receptors are equally permeable to Na+ and K+ but have a particularly high permeability to the divalent cation, Ca2+, unlike the pentameric excitatory channels. NMDA receptors are the site of action of ketamine, nitrous oxide and xenon, all of which are non-competitive inhibitors of glutamate. There is a high density of NMDA receptors in the hippocampus and associated regions, which are important in the formation and recall of memories.
Ionotropic purinergic receptors: P2X subtypes
Receptors of this family form cation channels that are equally permeable to Na+ and K+ but also to Ca2+. They are activated by ATP and its metabolites and are widely distributed in both central and peripheral neurones. The analgesic property of pentobarbital is thought to be due to inhibition of P2X receptors in the dorsal root ganglia. These ionotropic purinergic receptors are not to be confused with G-protein-coupled receptor forms of purinergic receptors: adenosine receptors (P1) and P2Y subtypes.
G-protein-coupled receptors (GPCRs)
Almost 1000 genes for GPCRs have been identified and all have a similar structure, although they can be divided into several distinct families. GPCRs have seven helical TMDs, starting with the extracellular N-terminus and ending with the intracellular C-terminus. The quaternary structure is such that these helices cluster together, held in the correct alignment by interactions with extracellular/intracellular domains. When a ligand binds, these helices twist in relation to each other, inducing a conformational change that is transmitted to the domain that is associated with G-protein coupling. The ligand binding site is determined by the composition of the second and third extracellular loops, whereas the second and third intracellular loops are associated with G-protein binding.
All adrenoceptors, muscarinic, cholinergic and opioid receptors work through a GPCR mechanism. G proteins are associated with the inner leaflet of the cell membrane and are not always associated with receptors. When the ligand binds, a conformational change occurs that increases the likelihood of the receptor becoming associated with its particular G protein. The kinetics of GPCR–G-protein interactions requires complex models to explain observed responses. Essentially the receptor can exist in a number of states, which differ in their affinity for agonist, antagonist or inverse agonist. Response is greatest when a full agonist is bound to the appropriate G protein. Once the GPCR–G-protein association takes place, there is a conformational change in the G-protein α subunit that allows dissociation of bound GDP in exchange for GTP and favours dissociation of the α subunit from the βγ dimer. The GTP-bound α subunit now has sufficient energy to interact with intracellular enzymes or cell-membrane-bound ion channels and either activate or inhibit these secondary mechanisms. The GTPase activity of the α subunit limits the duration of this activity and, once GTP is hydrolysed to GDP, further interaction is energetically unfavourable and the α subunit then re-associates with the βγ dimer, which has dissociated from the GPCR. In some situations the βγ dimer can also act as an intermediary by opening K+ channels.
There are many different types of G protein, with subfamilies classified according to their α-subunit activity including Gs, Gi and Gq subfamilies: Gs and Gi proteins activate and inhibit adenylyl cyclase, respectively; Gq causes phospholipase C to hydrolyse phosphatidylinositol in the cell membrane, producing diacylglycerol (DAG) and inositol triphosphate (IP3). All catecholamine action is mediated through GPCRs, although different adrenoceptor subtypes are associated with different subfamilies of G protein. Figure 28.3 lists some GPCRs and their G-protein associations that are of importance to the anaesthetist.
| Natural ligand/receptor type | G-protein α-subunit type | Agonist/antagonist drugs |
|---|---|---|
| Acetylcholine M1, M3 and M5 | Gq | Atropine, glycopyrrolate antagonists |
| Acetylcholine M2 and M4 | Gi | Atropine, ipratropium, glycopyrrolate antagonists |
| Noradrenaline α1 | Gq | Phenylephrine agonist; phentolamine antagonist |
| Noradrenaline α2 | Gi | Clonidine agonist; yohimbine antagonist |
| Noradrenaline β1 and β2 | Gs | Isoprenaline, salbutamol agonist (β2); atenolol, propranolol, labetolol antagonist |
| Opioid receptors (all types) | Gi | Morphine, fentanyl, alfentanil, remifentanil (μ); pentazocine (κ) |
| GABAB receptors | Gi | Baclofen agonist |
| P1 adenosine receptors | Gi | Adenosine agonist |
| P2Y1 and P2Y2 receptors | Gi | ADP agonist; clopidogrel irreversible antagonist |
| Histamine H1 receptors | Gq | Cetirizine antagonist |
| Histamine H2 receptors | Gs | Ranitidine antagonist |
| Dopamine D1 and D5 receptors (postsynaptic) | Gs | Dopamine and dobutamine in kidney agonist |
| Dopamine D2, D3 and D4 receptors (presynaptic) | Gi | Bromocriptine agonist; haloperidol, risperidone, chlorpromazine and clozapine (D4 selective) antagonists |
| Serotonin 5-HT1A receptors | Gi | Buspirone antagonist |
| Serotonin 5-HT2 receptors | Gs | Ketanserin antagonist |
| Angiotensin II AT1 receptors | Gq | Losartan, valsartan antagonist |
Other transmembrane messenger systems
Unlike GPCRs, tyrosine kinase receptors (Trk) do not rely on an intermediary protein for activity. The cytoplasmic portion of a Trk forms the catalytic site, which is activated by ligand binding to the extracellular portion of the receptor. The effects of many polypeptide growth factors, cytokines and hormones, including insulin, are through Trk activation.
The guanylyl-cyclase-coupled receptor (GCCR) is a single transmembrane protein responsible for the action of certain hormones, particularly atrial natriuretic peptide.
Voltage-gated ion channels
Voltage-gated ion channels are activated by a change in membrane potential. They are present in nerve axons, including presynaptically, and on smooth and skeletal muscle.
Local anaesthetics such as lidocaine and bupivacaine act by blocking voltage-gated sodium channels. They are usually administered in close proximity to peripheral neurones; activity requires access to the cytosolic side of the axon so lipid solubility is important, although the ionised form is active. Some anticonvulsants, such as phenytoin, lamotrigine and carbamazepine, act by blockade of central sodium channels, so reducing neuronal excitability. Voltage-gated calcium channel blockers such as nifedipine and verapamil act by blocking L-type calcium channels: antihypertensive and antianginal effects are associated with action on vascular smooth muscle, whereas myocardial effects produce their antiarrhythmic action.
Intracellular receptors
Receptors within the cell can be associated either with the cytosol or with any of the specialised intracellular membranes; of particular importance are receptors associated with the sarcoplasmic reticulum that regulate calcium release.
Intracellular hormone receptors
Lipid-soluble hormones interact with intracellular receptors. There is a superfamily of such receptors, including those for sex hormones, corticosteroids, thyroxine and vitamin D3. These receptors act as ligand-regulated transcription factors that bind to DNA and influence the pattern of RNA production by either increasing or inhibiting specific protein production. Gene transcription is influenced by the recruitment of additional proteins that act as co-activators or co-repressors to remodel the quaternary structure of DNA. The oestrogen-receptor modulator tamoxifen inhibits transcription associated with tumour cells in certain cancers. In addition to hormone receptors, other nuclear receptors can also influence protein production: the antidiabetic drug rosiglitazone is a peroxisome proliferator-activated receptor type γ (PPAR-γ) agonist that stimulates protein transcription, leading to insulin sensitising activity within adipose tissue.
Adrenal steroid hormones
There are two types of corticosteroid receptor: MR (or type 1) is the mineralocorticoid receptor and GR (or type 2) the glucocorticoid receptor. In the cytoplasm, GR is bound to an inhibitory protein, heat shock protein. Glucocorticoid binding displaces this inhibitor and triggers a conformational change that facilitates translocation and binding to specific regions on DNA. GR is widespread in cells, whereas MR is restricted to epithelial tissue such as renal collecting tubules. Cortisol and aldosterone are equipotent at MR receptors: aldosterone-triggered activation is facilitated by the presence of 11β-hydroxysteroid dehydrogenase in epithelial cells, which metabolises cortisol to a compound that is inactive at the MR receptor.
Related posts:






Full access? Get Clinical Tree


