Mechanical Ventilation in Neuromuscular Disease: Introduction
The earliest application of assisted ventilation in patients with neuromuscular disease was with the iron lung during the poliomyelitis epidemic in the 1940s and 1950s, which enabled patients with severe respiratory insufficiency to recover, become ventilator-free, and go on to lead productive lives.1 In most cases, the device was used for a few weeks to up to 2 years, with eventual recovery of ventilatory function. Many polio patients with disability continue to reside in iron lungs in the community after several decades.2 Others have converted to tracheostomy-assisted positive-pressure ventilation, achieving mobility and the ability to clear airway secretions. With appropriate weaning techniques, some have switched to noninvasive positive-pressure ventilation (NIPPV).3 NIPPV is now preferred to support most patients with chronic neuromuscular disorders and is used increasingly to support patients with acute ventilatory insufficiency, such as with Guillain-Barré syndrome and myasthenia gravis. Table 32-1 lists neuromuscular conditions associated with respiratory impairment and failure.
|
Overview of Pertinent Pathophysiology
The primary muscle of inspiration is the diaphragm, which is innervated by the third through fifth cervical nerves. In addition, the external intercostal muscles provide stability to the rib cage during inspiration. With ascending paralysis, such as with Guillain-Barré syndrome or traumatic quadriplegia above the T10 level (Fig. 32-1),4 there are reductions in vital capacity and other volume subdivisions, as well as rib cage distortion with inspiratory effort. These changes result in regional ventilation–perfusion mismatching, particularly in the dependent portions of the lungs. An early increase in the alveolar-arterial oxygen (O2) difference can be found in some patients with neuromuscular impairment long before hypercapnia develops.5,6 Gas exchange worsens during sleep secondary to alveolar hypoventilation, inhibited intercostal and accessory muscle activity, and dead space ventilation induced by rapid shallow breathing.6–8 Long-term episodic hypoxia in dystrophic mice leads to dysfunction of myofibrillar contractility as there is no change in diaphragmatic collagen content and dry-to-wet ratio.9 Expiratory muscles in muscular dystrophies are more impaired than inspiratory muscles.10 Despite this finding, cough effort can still be preserved if inspiratory volume and respiratory elastic recoil are maintained.11
Figure 32-1
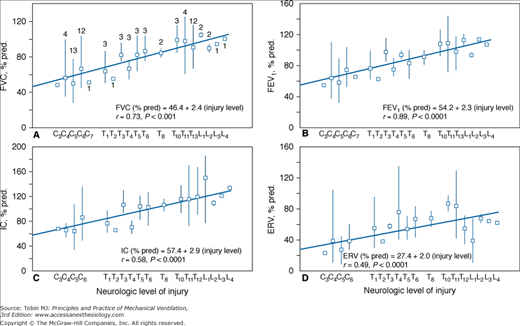
Spirometric variables in seated spinal cord-injured subjects distributed according to lesion level. A. Forced vital capacity (FVC; n = 74). B. Forced expiratory volume in 1 second (FEV1; n = 74). C. Inspiratory capacity (IC; n = 73). D. Expiratory reserve volume (ERV; n = 73). Values are means and ranges of percent predicted (% pred) values for healthy persons. Numbers above squares in A are numbers of subjects at each injury level and are the same for B to D. (Used, with permission, from Baydur et al.4)
Patients with Duchenne muscular dystrophy with a vital capacity above 1.8 L remain normocapnic.12 Below this value, nocturnal hypercapnia is likely to ensue. Many factors contribute to hypercapnia including respiratory muscle weakness,13 central hypopneas during rapid-eye movement sleep,14 upper airways obstruction,15 sleep fragmentation,16 obesity,14 respiratory muscle asynchrony,17 and decrease in respiratory compliance.18,19 Thoracic scoliosis, often associated with disorders such as poliomyelitis and muscular dystrophy (Fig. 32-2), further reduces respiratory compliance, resulting in an increase in inspiratory drive, altered force-length and force-velocity relationships,20,21 and, ultimately, diaphragmatic fatigue.
Body position influences regional ventilation in individuals with neuromuscular disease. Furthermore, use of positive-pressure ventilation and an abdominal belt results in greater global and regional ventilation in the supine position than in the right and left lateral positions.22 Intermittent positive-pressure breathing sessions (30 consecutive deep breaths at pressures up to 30 cm H2O) improve regional ventilation by approximately 30%, more so in the anterior regions.22,23
Measurement of maximal static mouth pressures (Pmax) provides a guide to the degree of respiratory muscle weakness and is an adjunct in assessing the need for assisted ventilation. In stable neuromuscular disease, maximal expiratory mouth pressures are lower in myopathy than in polyneuropathy and in proximal than in distal muscle weakness.10,24
Weakened abdominal muscles reduce the ability to cough, as reflected by decreased peak expiratory flow rates. Patients with peak expiratory flows of less than 160 L/min need cough assistance to prevent accumulation of airway secretions.25,26 Cough impairment is worsened by inspiratory muscle weakness and glottic dysfunction. Bulbar muscle involvement impairs swallowing and speech and increases risk of aspiration and upper airway collapse.
Dysregulation of breathing in muscular dystrophy and myotonic dystrophy has been attributed to abnormal feedback from respiratory muscle receptors and abnormal central ventilatory control, respectively.27,28 Sleep-disordered breathing can be related to respiratory muscle weakness, chest wall deformities, obesity, craniofacial abnormalities, bulbar dysfunction, and abnormalities in control of ventilation.14,29 Loss of rib cage and bulbar muscle tone, particularly during rapid-eye movement sleep, results in hypoventilation, decreased inspiratory flow, fragmented sleep, and daytime symptoms, well described in myotonic dystrophy, diaphragmatic paralysis, Duchenne muscular dystrophy, and amyotrophic lateral sclerosis.30–34 Despite muscle weakness, central drive in patients with neuromuscular and chest wall disorders is often increased.35
Clinical and Physiologic Indications to Initiate Assisted Ventilation
Altered mental status, cardiac or respiratory arrest, shock, arrhythmias, gas-exchange abnormalities, and bulbar dysfunction are absolute indications for endotracheal intubation. Such acute changes are frequently seen in patients with Guillain-Barré syndrome, myasthenia gravis, and almost always in cervical spinal cord injury. Lawn and Wijdicks36 advocated the use of a pulmonary function score, the sum of the vital capacity, maximum inspiratory pressure (PImax), and maximum expiratory pressure (PEmax), to predict prolonged ventilation and the need for tracheostomy in patients with Guillain-Barré syndrome.
The patient developing slowly progressive respiratory impairment will experience dyspnea, increased use of accessory neck muscles, paradoxical breathing, cough after swallowing (indicative of aspiration), tachycardia, sweating, and inability to say more than a few words in a row. Some patients may complain of headaches or sleep disturbances or exhibit cognitive impairment associated with daytime hypercapnia with partial compensation. Those with generalized muscle weakness often experience orthopnea whereas patients with quadriplegia prefer the supine position because the diaphragm assumes a greater resting length and force generation.4
Serial measurements of forced vital capacity provide a simple and reliable means of assessing disease progression, and may predict difficulties with cough and secretion clearance37 although as much as 50% reduction in respiratory muscle strength occurs before these values are reduced.38 In general, patients with less than 10% of predicted (or <1 L) vital capacity have little tolerance off assisted ventilation.2,3 In patients with bulbar involvement who have difficulty holding a mouthpiece in place, a full face mask can be used to perform spirometry. Facemask measurements tend to produce vital capacity values larger than those recorded by mouthpiece, in some cases by as much as 1.2 L in the seated position.39 A more useful guide to diaphragmatic weakness is the comparison of the vital capacity measured in the seated and supine positions. Reductions in vital capacity of 12% to 65% have been recorded in patients with neuromuscular disease3,4,40,41 (except in those with traumatic quadriplegia4,42,43) on assuming the supine position.
Measurements of PImax and PEmax are noninvasive and have established normal values for adults and children. They require, however, maximal effort and coordination, and are uncomfortable to perform. Low values may be difficult to interpret.44,45 Poor effort or air leaks around the mouthpiece give rise to submaximal measurements. Sniff esophageal pressure (Pes-sniff) and twitch transdiaphragmatic pressure (Pdi) tests are more precise but somewhat invasive (Fig. 32-3).46–48 Sniff nasal inspiratory force avoids the need for use of a mouthpiece and is easily performed by most patients, but may be tiring because of the need for repetition.49,50 A sniff nasal inspiratory force less than 40 cm H2O2 has a sensitivity of 97% and specificity of 79% for predicting 6-month mortality in amyotrophic lateral sclerosis.51 The use of individual tests tends to overdiagnose respiratory muscle weakness by approximately 27%, while the combination of tests increases precision and reduces the diagnosis of respiratory muscle weakness by 19% to 56% (Fig. 32-4).52 Supine forced vital capacity, upright forced vital capacity, PImax, PEmax, and Pdi-sniff are associated with tracheostomy-free survival, more so than PaCO2 (Fig. 32-5).41
Figure 32-3
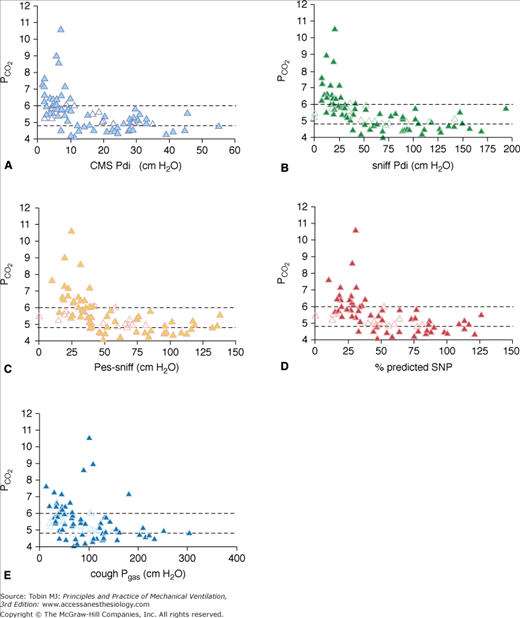
Relationship between tests of respiratory muscle strength (RMS) and PCO2. Each graph shows earlobe blood gas PCO2 in kPa on the y-axis and RMS on the x-axis. Open symbols represent the bulbar patients and closed symbols the limb patients. The dotted lines represent the lower and upper limits of normal for earlobe blood gas PCO2 (4.8 to 6.0 kPa). CMS Pdi, transdiaphragmatic pressure after bilateral cervical magnetic phrenic nerve stimulation; cough Pgas, cough gastric pressure; sniff Pdi, maximal sniff transdiaphragmatic pressure; Pes-sniff, maximal sniff esophageal pressure; SNP, maximal sniff nasal pressure. (Used, with permission, from Lyall A, Donaldson N, Polkey MI, Leigh PN, Moxham J. Respiratory muscle strength and ventilatory failure in amyotrophic lateral sclerosis. Brain. 2001;124:2000–2013, with permission of Oxford University Press.)
Figure 32-4
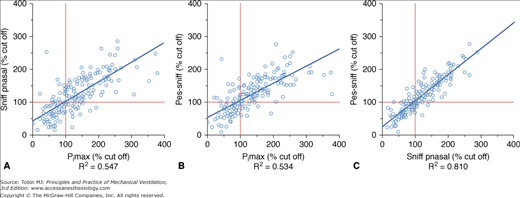
Correlation between (A) maximum inspiratory pressure (PImax) and sniff nasal pressure (Pnasal), (B) PImax and sniff esophageal pressure (Pes), and (C) Sniff Pnasal and Pes-sniff. (Reproduced from Steier J, Kaul S, Seymour J, Jolley C, Rafferty G, Man W, Luo YM, Roughton M, Polkey MI, Moxham J. The value of multiple tests of respiratory muscle strength. Thorax. 2007;62;975–980, copyright 2007 with permission from BMJ Publishing Group Ltd.)
Figure 32-5
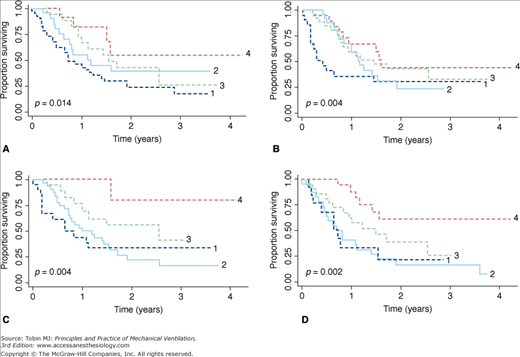
Kaplan-Meier curves showing differences in survival based on values of supine forced vital capacity (FVC) (A), upright FVC (B), maximal inspiratory pressure (PImax) (C), and maximal expiratory pressure (PEmax) (D). The categories of FVC are as follows: FVC ≤50% (line 1); FVC = 51% to 65% (line 2); FVC = 66% to 80% (line 3); FVC >80% (line 4). The PImax categories are as follows: PImax greater than −25 cm H2O (line 1); PImax = −25 to −50 cm H2O (line 2); PImax = −51 to −70 cm H2O (line 3); PImax less than −70 cm H2O (line 4). The PEmax categories are as follows: PEmax <25 cm H2O (line 1); PEmax = 25 to 50 cm H2O (line 2); PEmax = 51 to 70 cm H2O (line 3); PEmax >70 cm H2O (line 4). The p values compare differences in survival between the four categories of each pulmonary test by the Wilcoxon test. (Used, with permission, from Schmidt et al.41)
Table 32-2 lists physical and functional findings used in the evaluation of patients with impending respiratory failure secondary to neuromuscular disease.
|
Ventilator Targets in Neuromuscular Disease
The chief goals are to reverse altered mental status, correct gas exchange, and improve quality of life (see Table 32-2). With the use of nighttime NIPPV, daytime hypercapnia in Duchenne muscular dystrophy is delayed by 4 to 5 years53 and survival improves by 5 to 10 years.54,55 Application of a few hours of daytime NIPPV reduces breathlessness and increases endurance capacity in Duchenne muscular dystrophy more effectively than nocturnal NIPPV alone (Fig. 32-6).55 Similarly, NIPPV improves survival and quality of life in amyotrophic lateral sclerosis, even in those with bulbar dysfunction.56,57
Figure 32-6
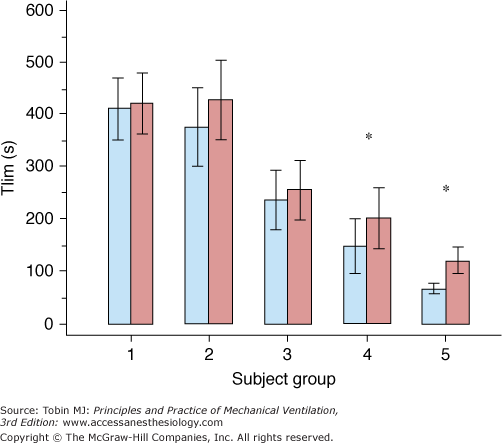
Endurance capacity (Tlim) in the five groups of patients with Duchenne muscular dystrophy. Columns represent mean values; errors bars represent 1 standard error of the mean (SEM). Blue columns: Evening Tlim; red columns: morning Tlim; *p < 0.05 (paired t test, evening vs. morning). Group 1, no assisted ventilation and 24/24 hours normocapnia; group 2, nocturnal hypercapnia before the start of noninvasive positive-pressure ventilation (NIPPV) implementation; group 3, nocturnal NIPPV with no breathlessness; group 4, nocturnal NIPPV with breathlessness; group 5, full-time NIPPV. (Used, with permission, from Toussaint M, Soudon P, Kinnear W. Effect of non-invasive ventilation on respiratory muscle loading and endurance in patients with Duchenne muscular dystrophy. Thorax. 2008;63:430–434. Adapted by permission from BMJ Publishing Group Limited.)
Experience with Traditional Modes of Ventilation
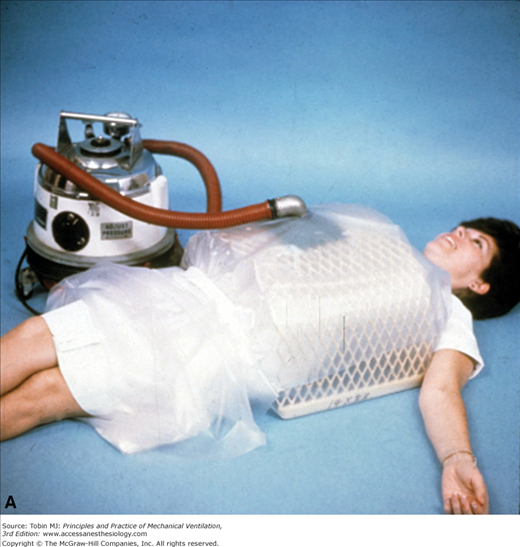
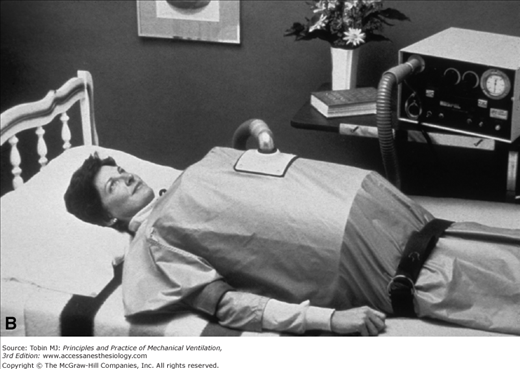
Many patients with poliomyelitis were supported with negative-pressure devices such as oscillating beds (Fig. 32-7), iron lungs (Fig. 32-8), cuirasses, and “ponchos” (Figs. 32-9A and B). Subsequently, individuals with other neuromuscular disorders received tracheostomies for positive-pressure support (Fig. 32-10). Disadvantages of tracheostomies include the necessity of teaching the patient and caregivers how to maintain the tracheostomy, and complications such as stomal infections, airway erosions (from repeated suctioning), tracheal stenosis, and difficulty with speech and swallowing (that can be overcome with proper speech and bulbar training and use of a one-way speaking valve). Airway-vascular fistulas (with fatal hemorrhage) and tracheobronchomalacia are infrequent but devastating complications that can occur even with uncuffed tracheostomies.58 Tracheostomies can be performed with local anesthesia during NIPPV support in hypercarbic patients with neuromuscular disease (vital capacity <20% predicted).59