Chapter 124 Malignant Hyperthermia
Pathophysiology
In fulminant MH, a dramatic increase in the metabolic rate of genetically abnormal muscle results in muscle injury and multiorgan system failure. The underlying biochemical defect is a sudden, sustained increase in the concentration of calcium ion in the sarcoplasm.1 Carbon dioxide (CO2) production increases severalfold. Even with increased minute ventilation, it may not be feasible to maintain normocarbia. Lactic acid production overwhelms the body’s buffering capacity. Increased O2 demand and the concomitant sympathetic response stress the cardiovascular system. High circulating catecholamines may stimulate muscle metabolism and hasten fulminant MH, but MH can occur in the presence of total sympathetic blockade and normal plasma catecholamines. Conversely, dantrolene, which can prevent MH in the susceptible patient, has no effect on stress-induced increases in catecholamines.2
MH can progress rapidly to severe mixed acidosis, hyperkalemia, elevated temperature as in heatstroke,3 and rhabdomyolysis. Renal failure, disseminated intravascular coagulation, cerebral edema, pulmonary edema, dysrhythmias, and cardiovascular collapse are potential consequences of fulminant MH. Even with aggressive treatment, death may ensue. Before dantrolene, the mortality rate of MH was 70%. Symptomatic therapy including mechanical ventilation, active cooling, administration of bicarbonate, expansion of the intravascular volume, and treatment of dysrhythmias can prolong life during an episode of fulminant MH. However, the most effective therapy is intravenous dantrolene.4
In the majority of MH-susceptible (MHS) humans, there is a defect in the ryanodine receptor channel (RYR1).5 Normally, depolarization of the sarcolemma is propagated through the transverse tubule system. There, the dihydropyridine receptor (DHPR), the skeletal muscle L-type calcium channel, undergoes conformational change in response to depolarization. This is coupled to opening of RYR1 through which calcium flows out of the sarcoplasmic reticulum (SR) into the myoplasm. The interaction between DHPR and RYR1 also activates entry of extracellular calcium (ECCE) into the myoplasm. The α2δ1 subunit of the DHPR is necessary for sustaining calcium transients in response to repeated action potentials.6 MHS RYR1 has increased sensitivity to agonists that open the channel7,8 and decreased sensitivity to inhibitors of RYR1 channel opening. Furthermore, ECCE is greater than normal in myotubes expressing MHS RYR1.9 Dantrolene inhibits ECCE in both MHS and normal muscle. Store-operated calcium entry (SOCE) is another process, occurring after depletion of SR calcium, that moves extracellular calcium into the myoplasm. SOCE is coupled to RYR1 and decreased by dantrolene.10
Genetics
MHS has been described as a syndrome with autosomal dominant inheritance, incomplete penetrance, and variable expressivity. The first-degree relatives of an individual that has had fulminant MH are considered potentially MHS until they have normal results of in vitro muscle contracture testing with halothane and caffeine (CHCT). Incomplete penetrance means that a person with a MHS mutation may not experience MH during the first or subsequent exposures to MH trigger agents. Multifactorial inheritance may be relevant.11,12 Variable expressivity means that clinical symptoms of MH vary from minor to fulminant depending on factors such as the anesthetic agents, genetics, and temperature.13 MHS episodes are more often observed in males.14,15
The primary genetic locus of MH (MHS1) is the ryanodine receptor gene (RYR1) on chromosome 19q13.1. Variants associated with MHS are found throughout RYR1. Fewer than 30 of these met the rigorous criteria (see www.emhg.org) that prove causation of MH. In families with an MH-causative RYR1 mutation, genetic analysis is a useful initial step in the diagnosis of MHS.16,17 See www.mhaus.org for addresses of clinical diagnostic testing laboratories in the United States. This pathologic test has been useful to document MH susceptibility in postmortem muscle.
Several different genetic abnormalities are associated with MH. A variant associated with MH susceptibility was found in the α-1 subunit of the DHPR gene (CACNA1S) on chromosome 1q32.18 This is the MHS 5 locus. It is associated with MH in only a few families as of 2009. Other loci associated with MH susceptibility, MHS 2, MHS 3, and MHS 6, have been identified on chromosomes 17q11-24,19 7q22-24, and 5p respectively. There are not obvious candidate genes at these sites other than the DHPR α2/δ1 subunit in 7q22-24, but extensive examination of MHS families has not identified a variant there. It has been suggested that two different genetic loci may be required to produce the MH phenotype in some families.11,20 Although factors that modify the expression of MHS are not completely determined, active cysteines contribute to redox modulation and nitrosylation of RYR1.21,22
Clinical Recognition of a Malignant Hyperthermia Episode in Humans
The initial signs of acute MH are nonspecific (Box 124-1).23 The first or only MH signs reported to the North American MH Registry in 286 cases were hypercarbia (38%), sinus tachycardia (31%), or masseter spasm (21%).14 Inappropriately elevated (>38.8° C) or rapidly increasing temperature was one of the first signs in 8.2% and the only initial sign in 3.9%.14
Box 124–1 Positive Findings Consistent with MH
However, high temperature was one of the first three signs in 63.5% of cases, with median maximum temperature of 39.1° C. The skin may be mottled and cyanotic and muscles rigid enough to extend the legs. Other signs include tachypnea, sweating, arrhythmias (ventricular tachycardia, ventricular fibrillation), dark urine, and excessive bleeding. Mixed respiratory and metabolic acidosis, hyperkalemia, myoglobinemia, myoglobinuria, and increased serum creatine kinase can be noted. However, rhabdomyolysis does not always occur during acute MH.24
MH episodes can be fulminant or abortive. In the operating room, MH can develop rapidly after the administration of succinylcholine in the presence of volatile anesthetic agents, or insidiously during a long anesthetic.25 When the nonspecific early signs of MH are noted during induction of anesthesia, the potent inhalation anesthetics should be discontinued. This patient is considered to be MH-susceptible until proven otherwise and this episode is called abortive MH. Early termination of inhalation anesthetic agents in the presence of abortive MH may allow spontaneous resolution of the syndrome. On the other hand, there have been cases in which only mild signs of abortive MH occurred intraoperatively, but renal failure, hyperthermia, and death occurred postoperatively with no explanation other than MH.26
In a retrospective Danish study of 386,250 anesthesias that occurred over 6.5 years in 87 hospitals, the incidence of fulminant MH was 1 in 250,000 general anesthesias.27 In this series, there were no cases of fulminant MH during regional anesthesia or “nontriggering” general anesthesia. When only anesthesias that included the administration of succinylcholine and potent inhalational anesthetic agents, such as halothane, isoflurane, and enflurane, were considered, the incidence of abortive MH was 1 in 4200 cases. In this study, abortive MH was defined as a masseter spasm or moderate changes in vital signs with slight metabolic or respiratory acidosis.27 These clinical signs are not specific for MH.28 No deaths occurred in the patients with abortive MH in this series.
The fiftyfold difference in incidence between fulminant and abortive MH can be explained by early termination of anesthesia in the presence of abortive MH with resolution of the syndrome, and perhaps by the fact that although the episode was classified as abortive MH, the patient may not in fact be MH-susceptible. Because MH is a potentially fatal condition that may progress rapidly, the anesthesiologist may be inclined to overdiagnose episodes of abortive MH.
In this retrospective series,27 there was no information about further anesthetic experience or other pathologic evaluation of MH susceptibility, specifically the results of in vitro caffeine-halothane contracture testing, in those patients who had experienced abortive MH.
A person may have fulminant MH during one anesthesia and no symptoms at all during other similar anesthesias. A patient who survived an episode of fulminant MH is considered capable of having another episode, although he or she may have undergone many anesthesias uneventfully before the first episode of MH. Larach et al. reported that 77 of 152 patients who experienced serious MH episodes had two or more prior unremarkable general anesthesias.14 Patients with the MH trait are often symptom-free until exposed to the most common triggering agents, the volatile anesthetic agents such as sevoflurane, isoflurane, desflurane, and halothane and the depolarizing neuromuscular blocking agent, succinylcholine. If the pharmacologic triggering agent is eliminated from the patient before the development of profoundly decreased pH in the muscle, rapidly increasing temperature, and shock, the metabolic abnormalities may resolve readily. This sequence of events could be termed abortive MH.
Potential Systemic Complications
Complications were noted in approximately 35% of 181 MH events reported to the North American MH Registry. These included changes in level of consciousness or coma in 9.4%, cardiac dysfunction in 9.4%, pulmonary edema in 8.4%, renal dysfunction in 7.3%, disseminated intravascular coagulation in 7.2%, and hepatic dysfunction in 5.6%.14 Disseminated intravascular coagulation was associated with a 50-fold increased likelihood of cardiac arrest and an 89-fold likelihood of death.29 The likelihood of any complication increased 2.9 times for every 2° C increase in maximum temperature and 1.6 times for every 30 minutes of time between the appearance of the first clinical sign of MH and the beginning of dantrolene administration.14 Other complications reported were compartment syndrome, stroke after cardiac arrest, bilateral brachial plexopathy, generalized muscle weakness, significant muscle loss, and prolonged intubation.14
Ventilatory failure may occur early in an episode of fulminant MH. During a MH episode, desaturation can be also secondary to increased oxygen extraction. The workload of the respiratory system may be further increased by the occurrence of pulmonary edema. Pulmonary edema may be the result of capillary leak. It may be worsened by impaired cardiac contractility in the presence of acidemia. Cardiac dysrhythmias may occur in the presence of marked electrolyte abnormalities. In older patients, there may be foci of myocardial fibrosis as well. It has been hypothesized that such areas of fibrosis are the result of subclinical episodes of MH that produced increased levels of circulating catecholamines.30 Cardiac contractility can be impaired.
Rhabdomyolysis will occur when the energy supply of the muscle is exhausted. Clinical manifestations include myalgias, swollen extremities, red-to-brown urine due to myoglobinuria, and elevated muscle enzymes. It is noteworthy that enzymes commonly elevated in the blood during hepatic injury including LDH and SGOT will also be released from muscle when creatine kinase (CK) is markedly elevated. Rhabdomyolysis can produce electrolyte imbalance (hyperkalemia, hyperphosphatemia, hypocalcemia), metabolic acidosis, severe hyperuricemia, acute renal failure,31 and compartment syndrome. Melli et al. reported that the incidence of acute renal failure associated with myoglobinuria was 46%.32 Massive rhabdomyolysis, producing CK of greater than 20,000 IU/L, may occur in patients with underlying muscular diseases not necessarily related to MH. Thus rhabdomyolysis in the absence of increased metabolic rate, hypercarbia, and metabolic acidosis should not be assumed to be MH. However, the same treatment, including administration of dantrolene acutely, may be helpful to the myopathic patient who experiences rhabdomyolysis in the ICU. It will be helpful to the patient and his or her family if the underlying cause of rhabdomyolysis is determined, because the implications for medical care of relatives differ depending on the pathologic diagnosis (e.g., dystrophinopathy vs. MH).
Hyperthermia in the Pediatric Intensive Care Unit
In the PICU, the most likely cause of fever is a bacterial infection,33 but elevated temperature can also be the result of trauma, viral infection, lymphoma, and leukemia. Inadequate fluid replacement predisposes to increased core temperature in children.34 Excessive environmental heat with inadequate opportunity for evaporative heat loss can result in temperature elevation. In some cases increased core temperature can be associated with tachycardia, increased expired CO2, and metabolic acidosis, which are consistent with the expected increase in metabolic demand produced by fever. This can be so extreme as to mimic MH.
When hyperthermia occurs in a child with history of exposure to succinylcholine or volatile anesthetic agents, MH should be considered in the differential diagnosis. In a retrospective cohort study, conducted to analyze and identify the causes of hyperthermia in the PICU over a 9-year period, Schleelein et al. noted that the incidence of clinically diagnosed MH was low (0.4%).35 These cases were classified as “definite “ or “probable” MH23 by a Malignant Hyperthermia Association of the United States (MHAUS) hotline consultant.35 No information about CHCT or other pathologic tests that could support the diagnosis of MH for these patients is available.
Postoperative Fever
Postoperative fever (>38° C, >100.4° F) is common after surgery and usually resolves spontaneously. In establishing a differential diagnosis, it is very helpful to consider the timing of fever onset: immediate, acute, subacute, or delayed. The causes may be infectious or noninfectious. The most common noninfectious cause is a medication reaction, followed by blood transfusion and by trauma suffered prior to surgery or as part of surgery. The fever due to MH usually starts within 30 minutes after administration of the triggering agent, but has also been reported up to several hours later, after the anesthesia was discontinued.
Some clinical states, such as the increased temperature that accompanies improved circulation after cardiac surgery, share some of the features of MH.36 These situations may even include rhabdomyolysis, but it is the result of muscular injury from impaired circulation, not usually from a primary muscular disease.
Abortive Malignant Hyperthermia Episodes and Isolated Masseter Spasm
There is controversy about evaluation of the individual who has experienced abortive MH or isolated masseter spasm. Abortive MH could be relatively slowly evolving MH or a group of signs produced by processes completely unrelated to MH. Increased stiffness of the masseter muscles, even to the degree that endotracheal intubation is precluded, can be a normal response to succinylcholine.37–39 Because MH could develop some minutes to hours after an occurrence of masseter spasm, some anesthesiologists recommend cancellation of an anesthesia after such an occurrence and evaluation of the CK levels at 12 and 24 hours after the incident. Neurologic evaluation and muscle biopsy to rule out susceptibility to MH have also been recommended. Neurologic evaluation is relevant after abnormal intraoperative muscle tension because myotonia may be the underlying diagnosis. A mutation in the muscle sodium channel has been noted in one family evaluated after severe rigidity followed succinylcholine administration.40 Others recommend discontinuing the triggering agents of MH, careful evaluation of acid-base status, and continuation of the anesthetic and surgical procedure if no further problems are identified. Urine should be checked for the presence of myoglobin. If masseter spasm occurs without significant metabolic or cardiovascular changes, it is unlikely to be followed by fulminant MH.41 Similarly, the low specificity of the contracture test for MH may produce many false-positive results in such patients.42
Rhabdomyolysis
Rhabdomyolysis is characterized by muscle necrosis and the release of intracellular muscle constituents including CK, myoglobin, calcium, and potassium. CK levels greater than five times baseline are a sensitive definition of rhabdomyolysis. Rhabdomyolysis can be due to inherited or acquired causes, and the severity of clinical consequences ranges from asymptomatic increase of serum muscle enzymes to life-threatening hyperkalemia. The most frequent causes are trauma, overexertion, immobilization, alcoholism, vascular insufficiency, and orthopedic surgery. Grand mal seizures, delirium tremens, psychotic agitation, and amphetamine overdose can lead to rhabdomyolysis in individuals with normal muscles.32 Some drugs such as HMG-CoA reductase inhibitors (statins) and colchicine are directly myotoxic.32 Rhabdomyolysis may occur in patients with metabolic myopathies such as carnitine palmitoyltransferase deficiency, myophosphorylase deficiency (PYGM) or McArdle disease, myoadenylate deaminase deficiency (AMPD1), mitochondrial myopathy, or malignant hyperthermia susceptibility. Occasionally, patients with structural myopathies can develop acute rhabdomyolysis after strenuous exercise, after exposure to potent inhalation anesthetics, after exposure to other myotoxic drugs, or after a viral infection.
Treatment of an Episode of Malignant Hyperthermia
Remove Trigger Agents
When an episode of MH is suspected, it is prudent to alter, as soon as possible, the anesthetic technique with elimination of all triggering agents. In the operating room, if potent inhalation anesthetic agents were administered, these should be discontinued immediately and a nontriggering anesthetic technique administered. High fresh gas flows of 10 L/min or more, or charcoal filters, are needed to eliminate residual potent inhalational anesthetic agents from modern anesthesia machines. If the physician suspects MH, and especially if blood gas analysis has proven the presence of significant respiratory and metabolic acidosis, dantrolene should be administered immediately and supportive treatment should be started as soon as possible (Box 124-2). Treatment of the life-threatening complications should not detract from the need to continue monitoring the metabolic status and the continued administration of dantrolene, in increasing doses if necessary, until the metabolic state is normal. Resolution of some of these complications may take longer than the adequate treatment of the acute episode of MH.
Box 124–2 Management of an Acute MH Episode in the ICU
Administer Dantrolene
Dantrolene, a hydantoin with muscle relaxant properties, has greatly changed the treatment of and risk of death from MH. Before the introduction of dantrolene, Brit and Kalow reported a 36% MH survival rate with symptomatic treatment only.43 It was thought that dantrolene decreased calcium release from the SR by decreasing the mobility of a calcium ionophore that transports calcium across membranes. Although dantrolene interacts with amino acids 590-609 in the N-terminal fragment of the RYR144, its analog, azumolene, does not alter calcium release from the SR. Dantrolene does decrease both excitation-coupled calcium entry into muscle cells and store-operated calcium entry coupled to RYR1.9,10 It does not act on the neuromuscular junction or on the passive or active electrical properties of the surface membranes of muscle fibers. Therefore patients given effective doses of dantrolene have normal electromyograms and depressed force of muscle contraction.45
Dantrolene for intravenous administration (Dantrium) is supplied in 70 mL vials, containing 20 mg dantrolene sodium and 3 g mannitol. It must be diluted with 60 mL of sterile, preservative-free, distilled water. Dantrolene should be available in all locations where general anesthesia is administered. It should be immediately supplied to other areas of the hospital by the pharmacy. If dantrolene has to be obtained from a central location, such as the pharmacy, it must be stressed that the need for the initial and subsequent doses of drug is urgent. The initial dose of intravenous dantrolene for treatment of MH is 2.5 to 3 mg/kg.46 More than 10 mg/kg has sometimes been required to return metabolism to normal. As soon as dantrolene is ordered to treat fulminant MH, replacements should be obtained by the pharmacy. In children, the intravenous infusion of dantrolene, 2.4 mg/kg IV over 10 to 12 minutes, produced stable blood levels of about 3.5 μg/mL for 4 hours, after which a slow decline in plasma concentration occurred (Figure 124-1).47 It appears that the half-life of dantrolene in the plasma of children is somewhat shorter than in adults: 7 to 10 hours compared with 12 hours, respectively.45 This is consistent with the recommendation to repeat dantrolene (1 mg/kg) every 6 hours for prophylaxis against recurrence of MH in a child. Immediately after the administration of intravenous dantrolene, one may note a dramatic decrease in heart rate. This is the result of an effect on the underlying hypermetabolic state and is generally a reassuring sign that dantrolene is effectively controlling the episode of MH. However, a modest decrease in heart rate after the administration of intravenous dantrolene may be caused by increased intravascular volume. The other goals of the treatment with dantrolene are complete cessation of tachypnea and muscle rigidity, correction of hypercarbia, hyperthermia, electrolyte disturbances, and blood gas abnormalities. Also, urinary output should be increased and mental status should improve. A flow sheet recording heart rate and rhythm, arterial blood pressure, central venous pressure, minute ventilation, core temperature, urine output, mixed venous blood gas tensions, serum electrolytes, serum glucose, and total fluid intake is useful.
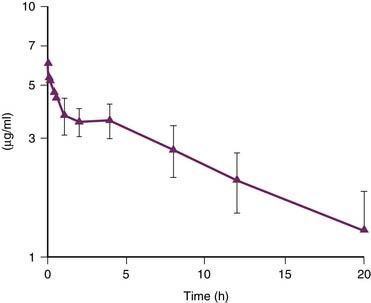
Figure 124–1 Whole blood concentration of dantrolene vs. time in a cohort of children.
(Modified from Lerman J, McLeod ME, Strong HA: Anesthesiology 70:625,1989.)






Full access? Get Clinical Tree


