CHAPTER 34
Lymphoma
Dana H. Manning, PharmD, RD, LDN
Hodgkin’s lymphoma (HL) and non-Hodgkin’s lymphoma (NHL) are neoplasms of lymphocytes and affect the anatomical sites where these cells develop and are processed, including the lymph nodes, the spleen, and the hematopoietic system. These cancers may spread to other tissues and structures, including the gastrointestinal tract, bone marrow, and liver. Lymphomas are considered cancers of the immune system, as they arise from the proliferation and accumulation of a mature single clone of lymphocytes (Gouveia, Siqueira, & Pereira, 2012).
Although the clinical presentation and initial diagnostic evaluation of these two diseases are similar, the diseases are distinct in that they arise from different malignant precursor cells and display unique genetic and pathophysiological characteristics. NHL is a clinically and biologically diverse group of diseases that may be divided into many subtypes; in contrast, HL is primarily one disease presentation with a small percentage of a different subset of the disease also described (nodular lymphocyte-predominant HL, which accounts for about 5% of the HL cases; Küppers, 2009). Although the disease presentations may be initially similar and the staging systems overlap, there is a great deal of difference in the most effective treatment regimen for each specific diagnosis. Fortunately, the development of less toxic therapies (some of which may be available as oral tablets) and better supportive care measures such as growth factors have lessened many of the complications of lymphoma treatment, enabling many patients with lymphoma to be treated as outpatients.
Primary providers participating in the care of these patients must understand the disease entities, the treatment modalities and their side effects, and complications. A multidisciplinary team approach is optimal to help decrease the morbidity and mortality rates associated with these hematologic malignancies. Primary care providers represent a vital piece in the care of patients with lymphoma, particularly in coordinating their cancer treatments with their chronic conditions and emphasizing evidence-based, effective, and safe supportive care of the patient’s symptoms and wellness.
 ANATOMY, PHYSIOLOGY, AND PATHOLOGY
ANATOMY, PHYSIOLOGY, AND PATHOLOGY
The intricate interrelationship between the hematologic and the immunologic systems protects the body from foreign invaders, such as pathogens and toxins. There are three primary components of the immune system. The first is phagocytosis, in which invading substances are ingested and destroyed by individual immune cells. Mononuclear phagocytes recognize invaders, engulf foreign particles, and activate specific immunity. The second component is inflammation, in which cells and proteins defend the body against infection and repair tissue damage. The third component of the immune response is adaptive immunity, mediated through the lymphocyte cells (B-cells, T-cells, and Natural Killer [NK] cells). These cells are critical to the recognition of self from nonself and the ability of the immune system to adapt and change. T lymphocytes are responsible for cellular immunity, whereas B-cells are responsible for humoral immunity through the production of antibodies. NK cells distinguish infected cells and tumors from normal and uninfected cells. Phagocytosis and inflammation are predominantly nonspecific immune responses, whereas humoral and cellular responses are characteristic of specific immunity.
Lymphocytes are formed from pluripotent stem cells, found primarily in the bone marrow. These cells differentiate into all of the recognized mature cell lines produced by the various hematopoietic cytokines, or growth factors (Owen, Punt, & Stranford, 2013). Figure 34.1 illustrates normal hematopoiesis and also notes the clinically relevant cytokines.
More than 1 billion lymphocytes are produced in the marrow daily. There are two functional classes of lymphocytes: the T lymphocytes, which regulate antibody synthesis and cellular immune processes; and the B lymphocytes, which contribute to the humoral response by producing specific antibodies to an identified antigen. The lymphoid stem cell is programmed by the bone marrow to develop into either a T-cell or a B-cell precursor. B-cell maturation occurs in the follicles of the lymph nodes after exposure to an antigen.
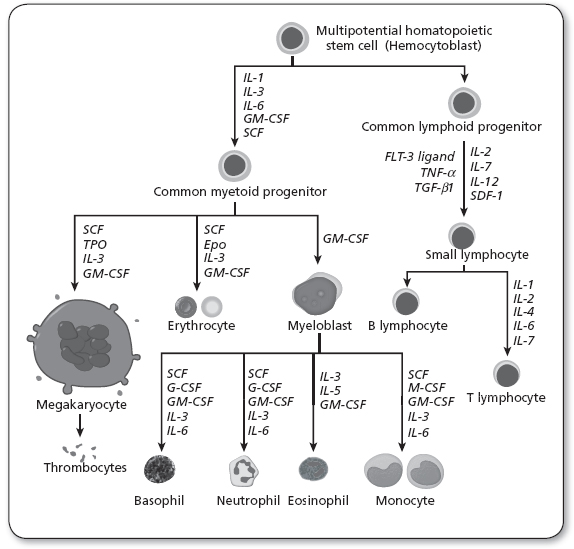
Figure 34.1
Diagram including some of the important cytokines that determine which type of blood cell will be created.
Epo, erythropoietin; FLT-3 ligand, FMS-like tyrosine kinase 3 ligand; G-CSF, granulocyte-colony stimulating factor; GM-CSF, granulocyte macrophage-colony stimulating factor; IL, interleukin; M-CSF, macrophage-colony stimulating factor; SCF, stem cell factor; Tpo, thrombopoietin; TNF-α, tumor necrosis factor-alpha; TGF-β, transforming growth factor beta; SDF-1, stromal cell-derived factor-1.
Lymphocytes are the only immunocompetent cells able to recognize antigens. T-cells are produced in the marrow, mature in the thymus, and then are released to secondary lymphoid organs, such as the lymph nodes, the spleen, the tonsils, and Peyer’s patches in the ileum, as well as throughout all the connective and epithelial tissues of the body. Lymphocytes continuously circulate through the lymphatic network and vascular channels throughout the body and within nodes, which are densely clustered in the neck, axillae, abdomen, and pelvis (Owen et al., 2013).
Surface markers and receptors, which are macromolecules with a binding affinity for a specific antigen, appear on the immune cell during maturation. They signal each other to recognize, bind, and kill antigens. Surface markers differentiate between lymphocyte subsets and are recognized by monoclonal antibodies. These groups of lymphocytes, surface markers, and monoclonal antibodies are called “clusters of differentiation” (CD). The most commonly known surface markers are CD4 and CD8. CD4 defines the helper function of T-cells, and CD8 the suppressor or cytotoxic function (Buettner & Bode, 2012; Owen et al., 2013).
When B lymphocytes are activated by a signal from T lymphocytes, they differentiate into one of the five classes of immunoglobulins (Ig); IgM, IgG, IgD, IgE, and IgA. In the primary immune response, there is a latent period in which B-cells make contact with the antigen, proliferate, differentiate, and secrete antibody. Because resting B-cells express only IgM and IgD, and IgD is rarely secreted, IgM is the dominant antibody secreted in the primary immune response. Other isotypes, including IgG, IgA, and IgE, appear with second exposure to the same antigen. The secondary antibody response is more rapid than the primary because of the added ability of memory. This complicated host response is distorted by malfunctions in hematopoiesis, especially the proliferation of abnormal cells. “Studies of cancer–immune system interactions have revealed that every known innate and adaptive immune effector mechanism participates in tumor recognition and control. The first few transformed cells are detected by NK cells through their encounter with specific ligands on tumor cells. This leads to the destruction of some transformed cells and the uptake and processing of their fragments by macrophages and dendritic cells. In turn, these macrophages and dendritic cells are activated to secrete many inflammatory cytokines and present tumor cell-derived molecules to T and B cells. Activation of T and B cells leads to the production of additional cytokines that further promote activation of innate immunity and support the expansion and production of tumor-specific T cells and antibodies, respectively. The full power of the adaptive immune system leads to the elimination of remaining tumor cells and, importantly, to the generation of immune memory to specific tumor components that will serve to prevent tumor recurrence (Finn, 2012, pp. 6–9).
Hodgkin’s Disease
Hodgkin’s disease (HD) was first described in 1832 by Thomas Hodgkin. The characteristic cellular changes of the disease were described approximately 70 years after that by Dorothy Reed and Carl Sternberg. These changed cells are the mononucleated Hodgkin’s cells and the multinucleated Reed–Sternberg (RS) cells. These cells, though pathognomonic for HD, may only account for 1% to 10% of the tumor tissue. The rest of the tumor cells may be a mix of other immunologic cell types (Küppers, 2009). HL is unique in that the changed cancer cells may have undergone reprogramming of gene expression to the extent that they lose many of their original characteristics (Küppers, 2009).
Classical HL almost always causes mass lesions, most typically in centriaxial lymph nodes, but occasionally in other organs, such as the spleen, bone marrow, liver, bone, or lungs. However, because several of these sites are anatomically deep within the body, indirect evidence of the presence of the lymphoma, such as constitutional symptoms, local organ-specific abnormalities (such as cough or bone pain), or incidentally discovered laboratory abnormalities, may precede detection of such mass lesions by physical examination or imaging (Connors, 2009).
Some lesions that originate in the mediastinum spread to nodes in the lower neck and then to the upper retroperitoneal area. Other histological types may skip the mediastinum and spread to the retroperitoneal nodes without ever causing intrathoracic disease. Disease spreads to extranodal areas by direct extension and then invades the vasculature, resulting in hematologic dissemination. The spleen is the final lymph node area of involvement before hematologous dissemination occurs. HL typically spreads in a predictable fashion from one set of lymph nodes to adjacent groups, most often starting in supradiaphragmatic nodes (90%) and much less commonly in infradiaphragmatic nodes (10%; Connors 2009; Banerjee, 2011).
Clinical Pearls
 Pain at sites of nodal disease, precipitated by drinking alcohol, occurs in <10% of patients but is a specific sign for HL.
Pain at sites of nodal disease, precipitated by drinking alcohol, occurs in <10% of patients but is a specific sign for HL.
Non-Hodgkin’s Lymphoma
Normally, the early lymphocyte develops in a predictable manner into a mature immunocompetent cell. However, in NHL, an abnormal proliferation of monoclonal cells occurs. Instead of progressing to the next developmental phase, the cells fix at one phase of development and continue to proliferate.
NHL is a large, heterogeneous group of malignancies, which originate from B lymphocytes, T lymphocytes, or NK cells. The largest proportion of cases involve the B-cells (80%–85%), with T-cell lymphoma accounting for the bulk of the remainder of the cases (15%–20%). NK-cell lymphomas are very rare (National Comprehensive Cancer Network [NCCN], 2013a). The neoplastic cell population is cloned from a single neoplastic hematopoietic stem cell and ultimately proliferates into lymphoma. Surface markers and immunoglobulins are formed after the proliferation of neoplastic cells and designate the cell type of lymphoma. This progression of events has important implications for the prognosis and treatment of this disease. B-cells can be divided into four cytologic types: small cleaved, large cleaved, small noncleaved, and large noncleaved. It is possible to predict some of the clinical manifestations of NHL based on the characteristics of the predominant cell type. Neoplastic cells often retain the surface of their cells of origin, so it is also possible to group them according to the phenotypic properties of their surface markers (Friedman, 2007). Classification of NHL is complicated and so is discussed fully in a separate section.
 EPIDEMIOLOGY
EPIDEMIOLOGY
With an incidence in excess of 61,000 cases a year, the malignant lymphomas collectively represent the sixth most common cause of cancer in the United States and account for approximately 4% of all new cancer diagnoses (Siegel, Naishadham, & Jemal, 2013). Approximately 85% of patients with malignant lymphomas have NHL, and the remaining 15% have HD. NHL accounted for approximately 3% of cancer deaths in 2012 (Seigel et al., 2013).
Hodgkin’s Disease
Each year, an estimated 9,290 patients are diagnosed with HL in the United States, and 1,180 people die from the disease. This makes it a relatively uncommon malignancy as compared to NHL and solid tumors. Most patients are diagnosed between 15 and 30 years of age, and there is another peak of diagnosis in patients 55 years of age and older (NCCN, 2013a, 2013b). HL is a relatively uncommon malignancy, with 7,000 to 7,500 new cases diagnosed annually in the United States of America. Most of these patients present with early-stage disease. The overall incidence of HL is lower in less-developed countries than in developed countries, with the exception of children younger than15 years, where a higher incidence is seen. There is only a mild increase in incidence throughout adolescence and young adulthood (Gobbi et al., 2013). Other etiologic differences are seen within the pool of HD patients as well. HD is more common in males than in females for all age groups, and half as common among African Americans as Whites.
HL is one of the few adult malignancies that can be cured in most instances. The salient feature of this lymphoma is the rarity (about 1%) of neoplastic elements in the cell population, whereas the overwhelming majority of cells are non-neoplastic, mostly consisting of T lymphocytes (Gobbi et al., 2013). Classic HL usually spreads by contiguity within the lymphatic tissue network, with a late extension to adjacent and distant viscera. Mortality from HL has been progressively decreasing, thanks to improving treatments and earlier diagnosis. This improvement has also been seen in younger patients as well. “Most children and adolescents with newly diagnosed high-risk Hodgkin lymphoma (HL) will achieve remission and cure with conventional chemotherapy with or without radiation therapy. However, these therapies can lead to long-term side effects (Nakatsuka, 2006). Therapy is titrated on the basis of risk group stratification using clinical prognostic factors and, in most cases, then refined through assessment of interim or end of chemotherapy response, primarily using functional imaging with fluoro-deoxyglucose positron emission tomography” (Kelly, 2012). Long-term side effects of therapies are one of the primary concerns of treating children who have lymphoma, and results in the differential treatment strategies even though the disease is clinically similar to that seen in adults.
Hodgkin’s Reed–Sternberg (HRS) cells can be characterized by lineage-specific markers, as well as those representing activation markers and transcription factors. In approximately 85% of cases, the HRS cells are positive for CD15 (the Lewis x blood group carbohydrate, 3-fucosyl N-acetyl lactosamine), whereas CD30 is expressed nearly universally. Most B-lineage associated markers are absent, but CD20 is expressed weakly in up to 50% of cases of classical HL (CHL; Blum, 2010). Certain human leukocyte antigen (HLA) phenotypes are associated with increased susceptibility, and there are several examples of multiple family occurrences. Familial HL represents 4.5% of all newly diagnosed cases. Anticipation (i.e., earlier onset and/or increasing severity in successive generations) occurs in families that exhibit both HL and NHL and it has been suggested that both neoplasms may have a common genetic basis. The relative risk of the development of HL increases approximately 100-fold in monozygous twins and sevenfold in siblings of patients younger than 45 years of age. However, the cumulative lifetime risks are very small for the development of HL de novo or in first-degree relatives of affected patients (Blum, 2010).
Modern molecular biology has added a great deal to the lexicon of prognostic factors available for most cancers. Numerous biologic markers have been identified as prognostic factors in HL, and include surface receptors, intracellular proteins, cytokines, and genetic abnormalities comprised of amplifications, deletions, epigenetic silencing, or alterations in microRNA in HRS cells or surrounding inflammatory cells (Blum, 2010).
Patients with a higher standard of living or who are better educated are also at increased risk. One hypothesis suggests that exposure to common environmental pathogens is protective against HD, and this exposure is reduced in wealthier young people. In older patients, social class characteristics may play a less significant role.
An association between Epstein–Barr virus (EBV) infection and HD has been postulated as a result of an increased incidence of HD in persons with a history of infectious mononucleosis. Prospective studies have also shown elevated EBV titers in patients before the diagnosis of HD. Twenty to 100% of HL occurrences appear to be associated with EBV infection, the association varying with age (more frequent in children and older adults), gender (more frequent in males), geography (higher in Asia than in the United States), and histology (more likely in MCCHL and LDCHL than in other subtypes). However, most adults who carry EBV do not go on to develop HL. It is possible that this association, which has not been fully elucidated, may be the result of the effects of viral oncogene activity. Fascinatingly, this association may go on to provide mechanistic targets for future therapies, including anticancer vaccination.
Non-Hodgkin’s Lymphoma
NHLs are a heterogeneous group of lymphoproliferative disorders originating in B lymphocytes, T lymphocytes, or NK cells. In the United States, B-cell lymphomas represent 80% to 85% of the cases, with 15% to 20% being T-cell lymphomas. NK-cell lymphomas are very rare. In 2013, 69,740 new cases of NHL and 19,020 deaths due to the disease were estimated; cases of chronic lymphocytic leukemia (CLL) are estimated separately (NCCN, 2013a). NHL was the seventh leading cause of new cancer cases in 2013, accounting for approximately 4% of the new cancer cases in both males and females. NHL is also the seventh and ninth leading cause of cancer deaths for females and males, respectively. These percentages work out to be approximately 70,000 new cases of NHL and 19,000 deaths from NHL in 2013 (Siegel et al., 2013). NHL is considered a family of lymphoid malignancies comprised of potentially 61 different disease entities that cause immune cell malignancies of B-cells and T-cells (Lymphoma.org). More than 90% of these malignancies considered under the classification of NHL are of B lymphocyte origin (Jaffe, 2009).
There has been a 50% increase in the incidence of NHL since the early 1970s, one of the largest increases for any cancer group, although this increase has moderated since the mid-90s (Lymphoma.org). This increase has been attributed partly to the HIV epidemic and the development of AIDS-related NHL. However, much of the increase in incidence has been observed in patients in their sixth and seventh decades; a large part of this increase incidence has paralleled a major decrease in mortality from other causes. The median age of individuals with NHL has risen in the last two decades. As a result, patients with NHL may also have significant comorbid conditions, which complicate treatment options (NCCN, 2013a). In the United States, the incidence of NHL shows a steady age-dependent increase from childhood through the eighth decade of life. Certain subcategories are more likely to occur in specific age groups. Diffuse large B-cell, lymphoblastic, and Burkitt’s lymphoma are the most common lymphomas in children. Aggressive lymphomas are the most common lymphoid neoplasms in young adults. Indolent lymphomas affect adults averaging age 56 years. Because the incidence of both indolent and aggressive lymphomas increases with age, they are the most common lymphoid malignancies in patients older than 60 years (NCCN, 2013a). Despite the challenges that present in particular patient populations, overall survival rates have increased for patients with an NHL diagnosis over the last three decades, from a 5-year survival of 47% for all races if diagnosed from 1975 to 1977 to a survival rate of 71% if diagnosed from 2002 to 2008 (Siegel et al., 2013).
NHLs are more common in males than females and in Whites than African Americans. Although NHLs are reported worldwide, specific subtypes occur more frequently in particular locales. The most well-established risk factor for the development of NHL is immunosuppression. Patients with HIV have an increased risk of developing high-grade NHL (Shankland, Armitage, & Hancock, 2012). It follows that locales with high rates of HIV will have correspondingly higher rates of NHL, such as some African countries. Others with immunological deficiencies, perhaps inborn or induced by immune-modifying medical treatment for other conditions, will also have an increased risk, although these patients are likely to be found at higher concentrations in modernized nations. In the Western world, the most common subtype of NHL is diffuse large B-cell lymphoma (DLBCL), which accounts for approximately 40% of all NHL cases (although DLBCL itself may be subdivided into morphological variants; Gouveia et al., 2012). Infection does play a part in development of some lymphomas, either by inhibition of immune function, or by other mechanisms, such as induction of chronic inflammatory response. The EBV, for example, has recognized associations with Burkitt’s and nasal NK-cell or T-cell lymphomas, and Helicobacter pylori infection is a risk factor for gastric mucosa-associated lymphoid tissue lymphoma (Shankland et al., 2012).
Some lymphomas have been associated with chromosome translocations and rearrangement of proto-oncogenes such as BCL-2, BCL-6, p53, and c-MYC. These changes may be important to the etiology and progression of the disease. In particular, the BCL-2 gene encodes for an anti-apoptotic protein that plays an important role in a mechanism of chemotherapy resistance; the BLC-6 gene codes for altered proteins, which may adversely affect cell differentiation and proliferation (Gouveia et al., 2012). The p53 gene is responsible for a more generalized mechanism of tumor promotion and functions in several different cancer types, as it plays an important role in DNA transcription, cell proliferation, and apoptosis. The MYC gene is more likely to be altered in individuals with AIDS and may predispose the individual to a more aggressive course. A similar process occurs in Burkitt’s lymphoma (associated with the immune-disrupting EBV), which is characterized by the dysregulation of MYC on chromosome 8 (Shankland et al., 2012).
Some research suggests that exposure to certain herbicides, industrial solvents, or vinyl chloride may increase a person’s risk. Other possible risk factors include exposure to excessive amounts of radiation or treatment with high-dose chemotherapy for cancer, or with immunosuppressive agents such as those used after solid-organ transplant, high-dose chemotherapy, or stem-cell transplantation (Shankland et al., 2012). NHL is 45 to 100 times more likely among renal transplant patients receiving immunosuppressive therapy, with lymphomas accounting for 29% of cancers in these patients. Other patients predisposed to NHL are those with autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus. Systematic reviews and meta-analyses of cohort studies indicate that autoimmune and chronic inflammatory conditions may increase the risk of developing NHL by standardized incidence ratios of 3.9 times (for rheumatoid arthritis) to 18.8 times (for Sjögren syndrome).
Certain viral etiologies have been implicated in NHL, including Epstein–Barr (particularly in the immunocompromised host), H. pylori, HTLV-1, and Kaposi sarcoma herpes virus. Burkitt’s lymphoma is associated with the presence of the EBV. The HTLV-1 virus has been implicated in some adult T-cell lymphomas, especially in those occurring in the Caribbean, parts of South Africa, and southwestern Japan. The HTLV-1 infection has also been detected in AIDS patients who develop lymphomas. “The effect of HIV varies by NHL subtype, with risks particularly increased for diffuse large B-cell lymphoma (30-fold), Burkitt lymphoma (50-fold), and central nervous system lymphoma (1,020-fold)” (Shiels et al., 2013).
 DIAGNOSTIC CRITERIA
DIAGNOSTIC CRITERIA
Lymphomas are diagnosed and categorized by the histological examination of an excised lymph node. This examination may include genetic analysis of the tumor cells as well as pathophysiological examination and the use of imaging studies to delineate tumor spread and organ involvement. Lymphomas are classified according to the World Health Organization (WHO) classification scheme, which was last modified in 2008 and included some important changes from the previous classification scheme. This scheme outlines the many different pathophysiological types of both NHL and HD, with an increasing influence of genetic characteristics on differentiating tumor classification. Both HD and NHL tumors are staged via the Ann Arbor system (Table 34.1). This staging does not account for changes in histology, bulky disease, or extranodal involvement, all of which are important prognostic indicators in NHL. The Cotswold additions, which address bulk and site of disease, make this system much more useful for staging NHL.
Lymphomas are classified by cell type, tumor characteristics, and genetic markers. The WHO classification represents a worldwide consensus on the diagnosis of lymphoma tumors and is based on the recognition of distinct disease utilizing specific diagnostic criteria (Figure 34.2). The characteristics of each of these disease entities are variable—the clinical courses may range from indolent to aggressive, and different genetic markers may be useful in defining the overall potential behavior of a tumor. These diagnostic classifications are continually evolving as new genomic and molecular markers are discovered within tumor types.
When diagnosing lymphomas, needle biopsy specimens are not adequate: The cell architecture must be preserved to make an accurate diagnosis. It is quite important that such a biopsy consist of either an entirely involved lymph node or a generous excisional biopsy of a deeper mass lesion to provide the pathologist with sufficient material to search for the expected RS cells and find the typical mixed inflammatory background changes. A definitive diagnosis is a cornerstone of modern oncologic management and should not be compromised by an inadequately small or crushed biopsy (Connors, 2009). Lymphomas without accessible lymph nodes may be diagnosed on the basis of bone marrow or peripheral blood smears using special stains and cytogenetics.
The histological diagnosis of HD is based on recognition of RS cells in the appropriate cellular and architectural setting. The RS cell is large, with two or more nuclei, each containing a single prominent nucleolus (Figure 34.3). A clear zone surrounding the nucleolus is a distinctive feature. However, cells similar to RS cells can be detected in other lymphomas and solid tumors, infectious mononucleosis, and lymphoid hyperplasia associated with phenytoin therapy. Therefore, the presence of RS cells can be a confusing finding.
In HD, the lymph node architecture is effaced by a cellular infiltrate composed of normal lymphocytes, eosinophils, histiocytes, and plasma cells, with RS cells scattered throughout this background of inflammatory cells. The accompanying lymphocytes are primarily host response and polyclonal T and B cells. Most of the T-cell population belongs to the T-helper subset.
Presently, HL is classified into two largely distinct entities, namely nodular lymphocyte predominance HL (NLPHL) and CHL, the latter being further subtyped as nodular sclerosis (NSCHL), lymphocyte rich (LRCHL), mixed cellularity (MCCHL), and lymphocyte depletion (LDCHL) subtypes. The lymphocyte-predominant, mixed cellularity, lymphocyte-depleted categories differ primarily in their relative proportion of RS cells, mononuclear variants, and reactive lymphocytes. Nodular sclerosis has broad collagen bands that divide lymphoid tissue into circumscribed nodules. Nodular sclerosis HD also has distinctive clinical features: it is the only HD subtype more common in women than in men, and it has a propensity to involve lower cervical, supraclavicular, and mediastinal lymph nodes.
Staging System for Lymphoma |
STAGE | ANN ARBOR STAGING SYSTEM | COTSWOLD MODIFICATION |
I | Involvement of a single lymph node region (I) or of a single extralymphatic organ or site (IE) | Involvement of a single lymph node region or lymphoid structure |
II | Involvement of two or more lymph node regions on the same side of the diaphragm alone (II) or with involvement of limited, contiguous extralymphatic organ or tissue (IIE) | Involvement of two or more lymph node regions on the same side of the diaphragm (the mediastinum is considered a single site, whereas the hilar lymph nodes are considered bilaterally); the number of anatomic sides should be indicated by a subscript (e.g., II3) |
III | Involvement of lymph node regions on both sides of the diaphragm (III), which may include the spleen (IIIs), a limited, contiguous extralymphatic organ or side (IIIE), or both (IIIES) | Involvement of lymph node regions on both sides of the diaphragm; III1 (with or without involvement of splenic, hilar, celiac, or portal nodes) and III2 (with involvement of paraaortic, iliac, and mesenteric nodes) |
IV | Multiple or disseminated foci of involvement of one or more extralymphatic organs or tissues, with or without lymphatic involvement | Involvement of one or more extranodal sites in addition to a site for which the designation E has been used |
Related posts:

Full access? Get Clinical Tree


