Figure 2.1. Disposition of sites for local anesthetics following peripheral nerve blocks. Nonspecific binding to extraneuronal tissues (e.g., tissue proteins, fat) and uptake into the blood stream limit the amount of drug available to produce neural blockade, and thereby affects the likelihood of adequate neural blockade. Placing the drug closer to the nerve decreases the impact of drug loss due to tissue and blood. Following uptake into the vascular system, some drugs are metabolized by plasma cholinesterases (e.g., chloroprocaine) or are delivered to the liver for metabolism (or both). Uptake into blood also plays a vital role in producing central nervous system (CNS) of cardiovascular toxicity.
B. Local anesthetic choice. In general, hydrophobic local anesthetics are cleared more slowly from an injection site than are hydrophilic drugs for the reasons noted earlier. In addition, hydrophobic drugs are intrinsically more potent than hydrophilic local anesthetics. Consequently, hydrophobic local anesthetics produce longer-duration blocks than do more hydrophilic drugs.
1. Vascular effects. Local anesthetics have a complex and variable effect on local blood vessels and consequently on their own clearance. In general, at high concentrations local anesthetics tend to produce vasodilatation, thereby increasing local blood flow and consequently their own clearance. As local anesthetic concentration falls, either as a function of distance from the injection site or because of clearance, vasoconstriction occurs thereby reducing clearance and prolonging duration (see specific drugs given in the subsequent text for individual differences).
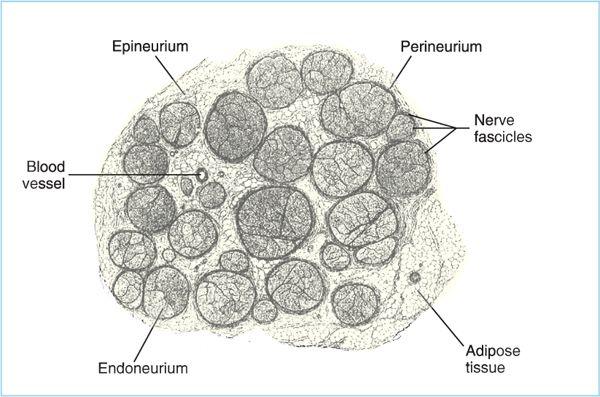
Figure 2.2. Drawing of a human peripheral nerve in cross section. Axons are gathered into fascicles, which contain dozens to a hundred or more axons. Each individual axon in the fascicle is surrounded by loose connective tissue (endoneurium). Each fascicle is surrounded by perineurium, and the collections of fascicles that make up the individual nerve are surrounded by epineurium. Perineurium and epineurium are more substantial barriers than is the endoneurium.
IV. Adjuncts
Multiple “adjuncts” are added to local anesthetics to alter their onset or duration. The most common follow:
A. Vasoconstrictors
1. Epinephrine added to a local anesthetic decreases local blood flow and, consequently drug clearance, thereby, prolonging block duration (2) and decreases local anesthetic peak plasma concentration following spinal, epidural, and peripheral nerve blocks. Lower peak plasma concentration decreases the risk of central nervous system (CNS) toxicity from systemic absorption and increases the local anesthetic dose that can be used safely. Importantly, epinephrine does not provide protection from the toxic effects of accidental intravascular local anesthetic injection (3).
a. The effect of epinephrine is generally greater for shorter acting, more hydrophilic drugs.
b. Most commonly, epinephrine is added to local anesthetics at a concentration of 5 μg/mL. There is insufficient data available to determine whether this is the optimal concentration for all local anesthetics and blocks.
c. The addition of epinephrine to 2-chloroprocaine for spinal anesthesia has been shown to produce “flu-like” symptoms (malaise, myalgias, arthralgia, anorexia) for unknown reasons (4). These symptoms do not occur when using plain 2-chloroprocaine for spinal anesthesia or when adding epinephrine to other local anesthetics for spinal anesthesia. Consequently, epinephrine should not be added to 2-chloroprocaine for spinal anesthesia. Fentanyl (20 μg) is an alternative that effectively prolongs spinal 2-chloroprocaine sensory block (5).
2. Phenylephrine has also been added to local anesthetics for spinal anesthesia. The dose is usually 1 to 2 mg (potency of 1 mg phenylephrine is equivalent to 0.1 mg epinephrine). However systemic absorption and subsequent hemodynamic effects when used in epidural or peripheral solutions limits its use. The dose is usually 1 to 2 mg (potency of 1 mg phenylephrine is equivalent to 0.1 mg epinephrine).
B. Clonidine
1. α2-Adrenergic agonists are analgesic drugs in their own right and have been shown to inhibit both C-fibers and A-fibers (6) and to modestly inhibit local anesthetic clearance (7). When added to local anesthetics, clonidine prolongs sensory block during peripheral, central neuraxial, and intravenous regional anesthesia to a degree comparable to that produced by epinephrine. However, unlike epinephrine, clonidine does not prolong motor block. Interestingly, clonidine is as effective at prolonging spinal block when administered orally as when added to the intrathecal local anesthetic (8).
2. The use of clonidine is limited by side effects, primarily sedation, hypotension, and bradycardia. These side effects result from the fact that presynaptic α2 adrenergic receptors inhibit norepinephrine release from noradrenergic neurons in the CNS.
C. Bicarbonate. Sodium bicarbonate (1 mEq/mL) is sometimes added to local anesthetics to increase speed of onset. However, studies are conflicting as to whether bicarbonate is effective in this regard. In general, bicarbonate does not speed the onset of hydrophobic local anesthetics (e.g., bupivacaine, etidocaine) and a positive effect with hydrophilic drugs has been demonstrated more often with epidural block than with peripheral nerve block. Even in those studies that have demonstrated a faster onset by adding bicarbonate, the effect is small and is of questionable clinical significance in most instances (i.e., it may take more time to locate and add the bicarbonate than is gained in faster onset). If bicarbonate is added to local anesthetics, care must be taken not to add too much (i.e., render the solution too alkaline) lest the local anesthetic precipitate. The most common recommendation is to add 1 mEq of bicarbonate to each 10 mL of lidocaine or mepivacaine, and one-tenth that amount to bupivacaine, if at all.
D. Hyaluronidase. This enzyme breaks down hyaluronic acid, which is an important component of connective tissue. It is added to local anesthetics in an effort to breakdown connective tissue in the extracellular matrix and thereby increase drug dispersion through tissue. It is of questionable clinical benefit and has been virtually abandoned for regional anesthesia except for peribulbar blocks. In peribulbar anesthesia the use of hyaluronidase is associated with a faster onset of motor block (9), but it has also been implicated in case reports of allergic reaction and injury to extraocular muscles.
E. Opioids
1. When added to short-duration local anesthetics used for spinal anesthesia, short-acting opioids (e.g., fentanyl, sufentanil) prolong and intensify sensory block without prolonging motor block or time to void (10), which is particularly advantageous for ambulatory spinal anesthesia. However, itching can be a problem (11). When added to local anesthetics for peripheral nerve block, fentanyl has also been shown to prolong sensory block, but at the expense of significantly slowing onset in some studies (12).
2. Systemic opioid effects/side effects cannot be ignored when they are added to local anesthetics. For example, when added to epidural local anesthetic infusions, fentanyl augments analgesia by systemic uptake and redistribution to brainstem, with all of the attendant risks of systemically administered opioids (13). Similarly, when added to intrathecal local anesthetics, the peak plasma concentration of sufentanil occurs between 20 and 30 minutes and is greater than what is necessary for postoperative analgesia (14). This explains the many reports of “early” respiratory depression in mothers (15) and fetal heart rate abnormalities in infants (16) when sufentanil is added to intrathecal local anesthetics for labor analgesia or cesarean (C)-section.
V. Differential block
Clinically, differential block refers to the observation that some nerve functions are blocked before others. This is probably a different phenomenon that occurs in vitro wherein different classes of nerve fibers are blocked at different local anesthetic concentrations.
A. In general, pain, autonomic function, temperature sensation (especially cold), and light touch are blocked before proprioception, deep pressure, and motor function.
B. Traditionally, this phenomenon was assumed to result from some nerve types having greater sensitivity to local anesthetic block. However, this explanation is at odds with in vitro studies showing that C-fibers (sympathetic, pain, temperature) are more resistant to local anesthetic block than Aδ-fibers (motor neurons) (see Chapter 1).
C. The mechanism responsible for differential block in vivo is still unknown but may involve the length of nerve exposed to local anesthetic, the position of the individual axons in the nerve bundle, the frequency at which the nerve fires, interference with neuronal firing pattern (which plays a role in the coding of sensory information projected centrally), and so on.
D. Differential blockade is more prominent with some local anesthetics, and is manifest by blockade of sensory propagation with apparent sparing of motor blockade at lower concentrations of drug. This sensory-motor dissociation is considered a beneficial effect for postoperative analgesia.
VI. Mantle effect
In addition to differential nerve block causing temporal differences in the sequence that various modalities are blocked, there are also spatial differences in block onset. That is, when blocking nerves that innervate an extremity, it is common to see that the more proximal parts of the extremity (i.e., those closer to the local anesthetic injection site) are blocked before more distal areas. This phenomenon is thought to result from the arrangement of axons within the nerve bundle such that nerves innervating distal parts of the extremity lie at the core of the nerve bundle, whereas those innervating more proximal portions lie in the mantle. Because local anesthetics move centripetally from the exterior of the nerve to the interior, the mantle fibers are blocked first.
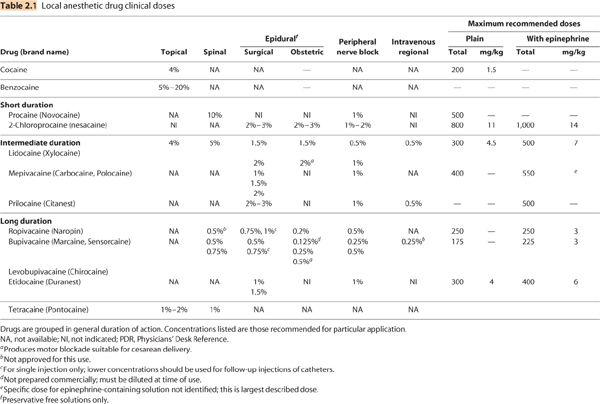
VII. Individual local anesthetics (Table 2.1)
A. Cocaine. Cocaine was the first local anesthetic used medicinally. It was abandoned as a local anesthetic for peripheral and central neuraxial blocks because of neural toxicity and abuse potential.
1. Cocaine is an ester local anesthetic metabolized in the liver to produce active metabolites. The half-life in humans is approximately 45 minutes. In the presence of alcohol, the metabolic pathway is altered to produce cocaethylene, which is more toxic than cocaine.
2. Currently, its “sole” medical use is as a topical local anesthetic (4%) in ear, nose, and throat (ENT) surgery because it produces intense vasoconstriction (thereby reducing bleeding) in addition to sensory block.
3. Maximum cocaine dose is 200 mg and because local anesthetic toxicity is additive, the common practice of performing awake nasal intubation with topical application of cocaine in the nose followed by liberal amounts of lidocaine (4%) or benzocaine spray to the pharynx/trachea increases the risk of systemic toxicity.
4. Cocaine can cause coronary artery spasm and concomitant use of cocaine and phenylephrine in nasal septoplasty has been associated with acute myocardial infarction in a 23-year-old patient without cardiac risk factors (17).
B. Benzocaine. Benzocaine was the first synthetic local anesthetic (although procaine was the first synthetic local anesthetic used clinically for nerve block).
1. It is an ester and a secondary amine with a pKa of 3.5. Consequently, it exists only in the uncharged form at physiological pH and is poorly soluble in aqueous solutions.
2. Because it is sparingly soluble in water, benzocaine is used exclusively as a topical spray, lozenge, or troche for mucous membranes or as a cutaneous cream/gel for dermal hypesthesia.
3. Although most local anesthetics have been implicated in causing methemoglobinemia, benzocaine appears to be particularly high risk in this regard. Adding to the inherent risk of methemaglobinemia is the fact that it is relatively easy to administer an excessive dose because of the difficulty in quantifying the amount of drug administered when it is applied as a spray or cream.
C. Procaine. Procaine is an ester and was the first synthetic local anesthetic used clinically.
1. Procaine is rapidly metabolized in plasma by cholinesterase and has an elimination half-life less than 8 minutes. Consequently, the risk of systemic toxicity is low.
2. Procaine is used primarily for subcutaneous infiltration (0.25%–1.0%). It is ineffective topically and is unreliable for epidural block. It is a poor choice for peripheral nerve block because of its slow onset and short duration.
3. Procaine is used for spinal anesthesia (50–100 mg). When compared to lidocaine, it produces a slightly shorter block and has a high failure rate (i.e., inadequate sensory block) but a significantly lower incidence of transient neurologic symptoms (TNS) (18). The commercial 10% solution should be diluted to 5% in dextrose, water, saline, or cerebrospinal fluid (CSF).
4. As with all synthetic ester local anesthetics, procaine is metabolized to para-aminobenzoic acid (PABA), which is a molecule frequently associated with allergic reactions.
D. Tetracaine. Tetracaine is the longest acting ester local anesthetic and before the advent of amide local anesthetics it was the preferred drug for long-lasting blocks.
1. As with procaine, slow onset when used for epidural or peripheral nerve block led to the abandonment of tetracaine for these uses when alternative amides were developed.
a. At one time, tetracaine was mixed with faster-onset local anesthetics (e.g., chloroprocaine) a failed effort to speed its onset while preserving its long duration (see Section VIII).
2. Tetracaine is metabolized (hydrolysis) more slowly than procaine (although it is still faster than the amide local anesthetics) are metabolized; consequently, risk of systemic toxicity is greater.
3. Tetracaine is used primarily for spinal anesthesia for which it is available as a 1% solution and as a powder (“niphanoid crystals”) that can be diluted with CSF, water, saline, or dextrose.
a. Tetracaine plus phenylephrine (5 mg) or epinephrine (0.2 mg) produces the longest-lasting spinal block (4–6 hours).
4. Tetracaine is very effective on mucosal membranes (commercially available in combination with benzocaine for this purpose) and is used for topical ophthalmologic anesthesia.
E. 2-Chloroprocaine. 2-Chloroprocaine, a derivative of procaine, was the last ester local anesthetic introduced into clinical practice.
1. Unlike procaine, 2-chloroprocaine has a rapid onset of action, and at concentrations of 2% to 3% is an effective drug for epidural, spinal, and peripheral nerve blocks. Because it has a duration of action between 30 and 60 minutes, it is suitable for short outpatient procedures.
2. 2-Chloroprocaine is hydrolyzed in plasma even more rapidly than procaine and has a half-life less then 1 minute. Therefore, the risk of systemic toxicity is lower than for any other local anesthetic.
a. The low risk of toxicity to mother and newborn plus its rapid onset makes chloroprocaine an attractive drug for epidural anesthesia for C-section.
3. Formulations. Chloroprocaine is available commercially as a preservative-free solution and as solution containing sodium bisulfite as an antioxidant (1). Because of concern regarding potential neurotoxicity, only the preservative-free solution should be used for central neuraxial block.
4. Use of 2-chloroprocaine for spinal anesthesia is controversial and is discussed in detail in Chapter 6. In brief, intrathecal 2% chloroprocaine, at a dose of 40 mg, produces good quality spinal anesthesia with a faster recovery and a lower incidence of TNS than lidocaine (19).
5. Interestingly, use of 2-chloroprocaine for epidural anesthesia has been shown to reduce the subsequent analgesic duration of epidural morphine, fentanyl, and clonidine. The mechanism is unknown.
F. Lidocaine. Lidocaine was the first amide local anesthetic introduced into clinical practice and it rapidly replaced the esters because of its longer duration and better quality block than procaine, its lower toxicity than tetracaine, and a much lower risk of allergy. It is the “archetypal” amide local anesthetic against which all others amides are compared.
1. Lidocaine is effective for peripheral nerve block (1% and 1.5%), epidural anesthesia (2%), spinal anesthesia (0.2%–5%), intravenous regional anesthesia (0.5%), and mucosal anesthesia (4%).
2. Lidocaine produces moderate vasodilatation.
3. Lidocaine is the local anesthetic most likely to cause TNS and while all local anesthetics can cause spinal cord injury, lidocaine may well be one of the most dangerous agents in this regard (20).
4. Although very rare, lidocaine allergy has been reported (21).
G. Mepivacaine. Mepivacaine is a cyclic tertiary amine like ropivacaine and bupivacaine, but clinically it is similar to lidocaine. It differs chemically from bupivacaine and ropivacaine in that it has a methyl group as a substituent on the tertiary nitrogen.
1. Mepivacaine is useful for infiltration, epidural, spinal, and peripheral nerve blocks. It is not very effective topically.
2. Mepivacaine has a mild vasoconstricting effect, which may explain its approximately 25% longer duration than lidocaine.
3. Mepivacaine is poorly metabolized in the fetus and neonate and is probably not a good choice for epidural anesthesia/analgesia in obstetrics (22).
H. Prilocaine. Prilocaine is clinically similar to lidocaine and although not commercially available for regional anesthesia in the United States, it is used in other countries.
1. Prilocaine has a large volume of distribution and is the most rapidly metabolized amide local anesthetic. These pharmacokinetic properties have led some to consider it an ideal drug for intravenous regional anesthesia.
2. Prilocaine is used in local anesthetic creams for cutaneous anesthesia.
3. Prilocaine’s unique metabolite, o-toluidine, causes methemoglobinemia, which has limited the clinical acceptance of prilocaine.
I. Bupivacaine. Bupivacaine was the first long-acting amide local anesthetic. It has a butyl group on the tertiary nitrogen where mepivacaine has a methyl group. This substituent makes bupivacaine significantly more hydrophobic than mepivacaine (and lidocaine), slower in onset but of much longer duration.
1. Bupivacaine is used for infiltration (0.25%), spinal (0.5% and 0.75%), epidural (0.5% and 0.75%), and peripheral nerve blocks (0.375%–0.5%). It is less desirable for intravenous regional anesthesia because of its cardiovascular toxicity. Peripheral nerve blocks with bupivacaine often provide sensory block for 4 to 12 hours and on occasion 24 hours. This has made it a useful agent for outpatient regional anesthesia of the extremities when prolonged analgesia is desirable. When instilled intraperitoneally, bupivacaine provides effective analgesia following laparoscopic surgery (23).
2. In the epidural space, dilute concentrations of bupivacaine (0.1% or less) provide good sensory analgesia with little or no motor block. This has made it a popular choice for both postoperative and labor epidural analgesia.
3. Bupivacaine, like other hydrophobic amides, has a lower therapeutic index with respect to cardiovascular toxicity than lidocaine. High plasma concentrations required for cardiovascular toxicity are usually associated with intravascular injection. Because bupivacaine is more slowly absorbed into plasma than lidocaine, it produces peak plasma concentrations that are approximately 40% lower (mg/mL per 100 mg administered). Consequently, bupivacaine is less likely to cause systemic toxicity than lidocaine if intravascular injection is avoided.
4. Reports of cardiac arrest following intravascular bupivacaine administration during attempted epidural anesthesia using 0.75% bupivacaine in pregnant women led the U.S. Food and Drug Administration (FDA) to warn against the use of this concentration for obstetric epidural anesthesia.
J. Levobupivacaine. Bupivacaine exists as two enantiomers, (R) and (S). Commercial bupivacaine is a racemic mixture of both enantiomers, whereas levobupivacaine is the pure (S)-enantiomer. It is not available in the United States at this time.
1. Levobupivacaine is approximately equivalent to the racemic mixture (i.e., bupivacaine) with respect to its use in regional anesthesia.
2. Human volunteer and animal studies indicate that the CNS and cardiovascular toxicity of levobupivacaine is less than that of bupivacaine (24,25). From a practical point of view, this means that patients can be expected to tolerate a somewhat larger dose of levobupivacaine before experiencing cardiovascular collapse. However, levobupivacaine is still quite cardiotoxic if a sufficient dose is administered intravenously and care must be taken to prevent intravascular injection (e.g., test dose, incremental injection). Quoting Mather and Chang, levobupivacaine “… may be viewed as ‘safer’, but must not be viewed as ‘safe”’ (25).
K. Ropivacaine. Ropivacaine is part of the homologous series that includes bupivacaine and mepivacaine. Ropivacaine has an isopropyl group bound to the tertiary nitrogen in place of mepivacaine’s methyl group and bupivacaine’s butyl group. Like levobupivacaine, it is supplied commercially as a single enantiomer. It is available as 0.2%, 0.5%, 0.75%, and 1% solutions.
1. The potency of ropivacaine is suggested to be clinically equivalent to that of bupivacaine. However, that is probably an overly simplistic view. In truth, the relative potency of these two drugs differs depending on the system being studied. For example, Casati et al. demonstrated that the ED50 dose for femoral nerve block was the same for bupivacaine and ropivacaine. In contrast, both Polley et al. and Capogna et al. demonstrated ropivacaine was 40% less potent than bupivacaine when used in dilute solutions for epidural analgesia in labor (26,27). Similarly, Camorica et al. demonstrated that ropivacaine was approximately 35% less potent than bupivacaine when administered intrathecally for labor analgesia (28). In addition, studies comparing the two drugs for peripheral nerve block generally find that equivalent doses produce similar onset and quality of block, but bupivacaine has a significantly longer duration. Therefore, ropivacaine is probably not equipotent with bupivacaine on a milligram per milligram basis, at least not in all clinical situations. This should be kept in mind when comparing the drugs with respect to “motor-sparring” effects and cardiotoxicity.
2. Ropivacaine is clearly less cardiotoxic than bupivacaine on a milligram per milligram basis. However, when comparing equipotent doses the difference in toxicity is less clear. Therefore, as with levobupivacaine, ropivacaine should not be considered a “safe” local anesthetic whether it is “safer” than bupivacaine or not.
3. As with cardiovascular toxicity, myotoxicity of ropivacaine is less than that of bupivacaine on a milligram per milligram basis. It is unclear if myotoxicity is less when comparing equipotent doses.
4. Ropivacaine produces vasoconstriction at concentrations used clinically for nerve block. This likely explains why epinephrine has little effect on the duration of ropivacaine epidural or peripheral nerve block (29,30).
L. Etidocaine. Etidocaine is a derivative of lidocaine with an additional ethyl group on the intermediate chain and a longer aliphatic group on the tertiary amine. These chemical differences make etidocaine a very hydrophobic local anesthetic. It is commercially available outside the United States as 1%, 1.5%, or 2% solutions.
1. Etidocaine’s onset is similar to lidocaine but its duration is comparable to bupivacaine.
2. Etidocaine’s clinical potency is similar to that of mepivacaine with 1.5% solutions commonly used in the epidural space and 1% solutions used for peripheral nerve block.
3. Etidocaine is the only local anesthetic that blocks transmission in the spinal cord dorsal column during spinal anesthesia. It is tempting to attribute this to its greater lipid solubility resulting in more extensive partitioning into the myelin of sheaths of dorsal column neurons.
4. Etidocaine fell out of favor clinically because of its tendency to produce motor block that outlasted sensory block (lack of “sensory-motor dissociation”).
M. Articaine. Articaine is a structurally interesting local anesthetic that has a 5-membered thiophene ring instead of a benzene ring as the “hydrophobic tail.” It is classified as an amide because the thiophene ring is connected to the intermediate chain by an amide linkage, but it also has an ester side chain attached to the thiophene ring.
1. Articaine (4%) is used “exclusively” as a dental local anesthetic and has become the second most commonly used local anesthetic for dentistry in the United States since its introduction in 2000. Its popularity stems from its rapid onset, long duration, and lack of ester-related allergy risk.
VIII. Local anesthetic mixtures
Before the advent of modern amide local anesthetics, it was common to mix a rapid-onset but short-acting ester (e.g., procaine) with a slow-onset but long-acting drug (e.g., tetracaine). The goal was to produce a solution with both a rapid onset and a long duration. Mixing local anesthetics for this purpose is still practiced, particularly by surgeons. However, it is of questionable value at best. First, local anesthetic toxicity is additive, therefore the total dose of each local anesthetic must be reduced by half when they are mixed. Consequently, the total number of “fast-onset” and “long-acting” local anesthetic molecules present at the injection site will be half of what it would be if the drugs were used singly. Therefore, onset will be slower than usually results from the “rapid-onset” drug and will be shorter than that produced by the “long-acting” drug. For example, mixing chloroprocaine and bupivacaine will produce a mixture with onset and duration characteristics comparable to lidocaine. Consequently, mixing local anesthetics is a practice that should probably be relegated to historical interest.
IX. Depo local anesthetic preparations
The potential benefit of long-acting local anesthetic blocks without the need for catheters and pumps has driven an effort to produce depot-like preparations of currently available local anesthetics.
A. Animal models have shown the ability of multiple preparations, including gels, liposomes, polymer microspheres, and oil–water emulsions to produce long-duration local anesthetic blocks. To date, none of these preparations have come to market for parenteral use in humans. When they do, their benefit and their liability may well be the same. Specifically, unlike a catheter technique, local anesthetic administration cannot be “turned off” with a depo preparation; if the patient (or the physician) does not like the block they will simply have to “wait it out”—perhaps for days. Also, if the preparation produces toxicity, for example allergy, there will be no way to remove it quickly.
B. There are commercially available depo-preparations in the form of local anesthetic patches and creams intended for cutaneous anesthesia before dermatologic procedures or to treat cutaneous pain (e.g., “shingles”). These are reasonably effective but not without risk. Several cases of methemaglobinemia and CNS toxicity have been attributed to local anesthetic creams applied to children (31,32).
X. Summary
Local anesthetics differ from one another in terms of their onset, duration, relative sensory versus motor block, metabolism, and so on. In addition to their inherent properties, various adjuncts can be added to local anesthetics to change their clinical profile to meet the requirements of individual clinical situations. Awareness of these facts will allow clinicians to make rational local anesthetic/adjuvant selections to provide safe and effective regional anesthesia for their patients.
REFERENCES







Full access? Get Clinical Tree


