Intravenous Anesthetics
The concept of intravenous (IV) anesthesia has evolved from primarily induction of general anesthesia to total intravenous regional anesthesia (TIVA) (White PF, Eng MR. Intravenous anesthetics. In: Barash PG, Cullen BF, Stoelting RK, Cahalan MK, Ortega R, Stock MC, eds. Clinical Anesthesia. Philadelphia: Lippincott Williams & Wilkins; 2013:478–500). All IV anesthetics are sedative–hypnotics and produce dose-dependent central nervous system (CNS) depression (Fig. 18-1). Although propofol is both safe and effective, the “ideal” IV anesthetic is yet to be developed (Table 18-1).
I. General Pharmacology of Intravenous Anesthetics
Mechanism of Action
A widely accepted theory is that IV hypnotics (benzodiazepines, barbiturates, propofol, etomidate, ketamine) exert their primary effects through an interaction with the inhibitory neurotransmitter γ-aminobutyric acid (GABA) (Fig. 18-2).
Activation of the GABA–receptor complex increases transmembrane chloride conductance, resulting in hyperpolarization and functional inhibition of the postsynaptic neuron.
Benzodiazepines increase the efficiency of the coupling between GABA and its receptor (ceiling effect).
Barbiturates and propofol appear to decrease the dissociation of GABA from its receptor.
Ketamine’s CNS effects appear to be primarily related to its antagonistic activity at the N-methyl-d-aspartate (NMDA) receptor. In addition, ketamine inhibits neuronal sodium channels (modest local anesthetic activity) and calcium channels (cerebral vasodilation).
Dexmedetomidine is a centrally active α2-adrenergic agonist that produces potent sedative and analgesic-sparing properties and reduces sympathetic outflow from the CNS.
Pharmacokinetics and Metabolism
The rapid onset of the CNS effect of most IV anesthetics can be explained by their high lipid solubility and relatively high cerebral blood flow (CBF).
The pharmacokinetics of IV hypnotics are characterized by rapid distribution and subsequent redistribution into
several hypothetical compartments, followed by elimination (Table 18-2).
The primary mechanism for terminating the CNS effects of drugs used for the IV induction of anesthesia is redistribution from a central highly perfused compartment (the brain) to larger and well-perfused peripheral compartments (muscle, fat).
Most IV anesthetic agents are eliminated via hepatic metabolism (some metabolites are active) followed by renal excretion of more water-soluble metabolites.
For most drugs, the hepatic enzyme systems are not saturated at clinically relevant drug concentrations, and the rate of drug elimination decreases as an exponential function of the drug’s plasma concentration (first-order kinetics).
High steady-state plasma concentrations are achieved with prolonged infusions, hepatic enzyme systems can become saturated, and the elimination rate
becomes independent of the drug concentration (zero-order kinetics).
Perfusion-limited clearance describes hepatic clearance of drugs (etomidate, propofol, ketamine, midazolam) in which extraction largely depends on delivery to the liver. (Hepatic blood flow decreases with upper abdominal surgery and increasing age.)
The elimination half-time (T1/2β) is the time required for the plasma concentration to decrease 50%.
Wide variations in T1/2β reflect differences in volume of distribution (Vd), clearance, or both.
Careful titration of an anesthetic drug to achieve the desired clinical effect is necessary to avoid drug accumulation and the resultant prolonged CNS effects after the infusion has been discontinued.
Context-sensitive half-time is the time necessary for the plasma concentration to decrease 50% in relation to the duration of the infusion. (This is important for determining recovery after varying length infusions of sedative–hypnotic drugs.)
Many factors contribute to interpatient variability in the pharmacokinetics of IV sedative–hypnotic drugs (Table 18-3).
Pharmacodynamic Effects
The principal pharmacologic effect of IV anesthetics is to produce dose-dependent CNS depression (dose-response curves) manifesting as sedation and hypnosis (Fig. 18-3).
When steady-state plasma concentrations are achieved, it can be presumed that the plasma concentration is in equilibrium with the effect-site (receptor) concentration.
Efficacy of an IV anesthetic relates to the maximum effect that can be achieved with respect to some
measure of CNS function (e.g., benzodiazepines are less efficacious than barbiturates in depressing electrical activity in the brain).
Potency relates to the quantity of drug necessary to obtain the maximum CNS effect.
Most sedative–hypnotic drugs (except for ketamine) cause a proportional reduction in cerebral metabolism (CMRO2) and CBF, resulting in a decrease in intracranial pressure (ICP).
On the electroencephalogram (EEG), it is likely that whereas sedative doses produce activation of high-frequency activity, anesthetic doses produce a burst-suppression pattern.
Most sedative–hypnotic drugs can cause occasional EEG seizure-like activity despite also acting as anticonvulsants. One should differentiate between the epileptogenic activity (methohexital) and myoclonic-like phenomena (etomidate). Myoclonic activity is considered to be the result of an imbalance between excitatory and inhibitory subcortical centers owing to unequal degrees of suppression of these centers by the hypnotic drug.
Most sedative–hypnotics (the exception is ketamine) lower intraocular pressure in parallel with effects on ICP and blood pressure.
With the exception of ketamine (and to a lesser extent, etomidate), IV anesthetics produce dose-dependent depression of ventilation (transient apnea followed by decreased tidal volume).
Many different factors contribute to the hemodynamic changes associated with IV induction of anesthesia (Table 18-4).
The effects of IV anesthetics on neuroendocrine function are also influenced by surgical stimuli (release of vasopressin and catecholamines, decreased glucose tolerance).
Most IV sedative–hypnotic drugs lack intrinsic analgesic activity (except ketamine, which has pronounced analgesic-like activity).
Hypersensitivity (Allergic) Reactions
Allergic reactions to IV anesthetics or their solubilizing agents, although rare, can be life threatening.
With the exception of etomidate, all IV induction drugs have been alleged to cause some histamine release.
Although propofol does not normally trigger histamine release, life-threatening allergic reactions have been reported, especially in patients with a history of allergy to other drugs (most often muscle relaxants).
Barbiturates may precipitate episodes of acute intermittent porphyria in vulnerable patients. (The benzodiazepines, ketamine, etomidate, and propofol are reported to be safe.)
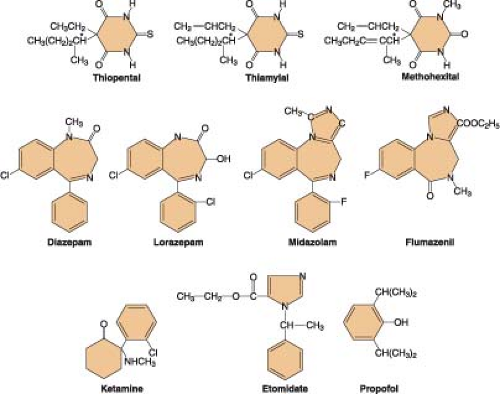 Figure 18-1. Chemical structures of nonopioid intravenous anesthetics. |
Table 18-1 Desirable Characteristics of an Intravenous Anesthetic | |
|---|---|
|
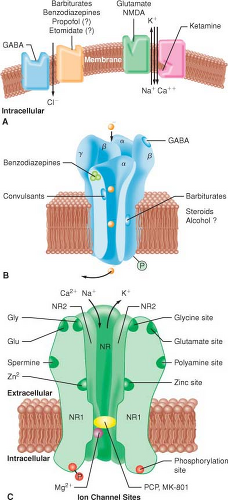 Figure 18-2. A model depicting the postsynaptic receptor sites for the inhibitory neurotransmitter γ-aminobutyric acid (GABA) and the excitatory neurotransmitter glutamate in the central nervous system. When GABA occupies the receptor, it allows inward flux of chloride ions, resulting in hyperpolarization of the cell membrane and subsequent resistance of the neurons to stimulation by excitatory neurotransmitters. Barbiturates, benzodiazepines, and possibly propofol and etomidate decrease neuronal excitability by enhancing the effect of GABA at this receptor complex. When glutamate occupies the binding site of N-methyl-D-aspartate (NMDA) subtype of the glutamate receptor, the channel opens and allows sodium, potassium, and calcium ions to enter or leave the cell. Flux of these ions leads to depolarization of the postsynaptic neuron and initiation of an action potential. Ketamine blocks these open channels, inhibiting the excitatory response to glutamate. |
Table 18-2 Pharmacokinetic Values for Intravenous Anesthetic Drugs | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|









