Chapter 82 Inotropes and vasopressors
The pharmacological support of the failing circulation is a fundamental part of critical care. The principal aim of these drugs is to restore inadequate systemic and regional perfusion to physiological levels.
DEFINITIONS
Inotropic agents are defined as drugs that act on the heart by increasing the velocity and force of myocardial fibre shortening. The consequent increase in contractility results in increased cardiac output and blood pressure. Characteristics of the ideal inotrope are shown in Table 82.1.
THE FAILING CIRCULATION
PHYSIOLOGY
Cardiac output is controlled by the peripheral vasculature that is as energetic at returning blood to the heart as the heart is at pumping blood to the periphery1 (Figure 82.1).
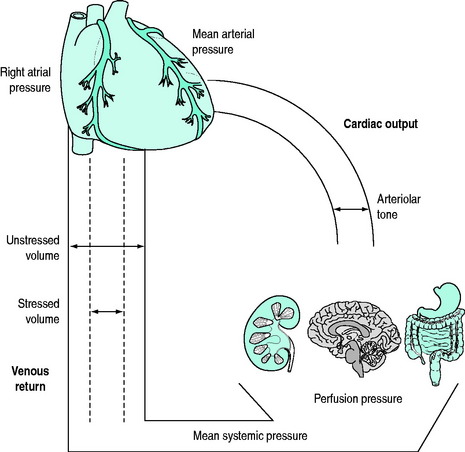
Under physiological conditions, the venous (‘capacitance’) system contains approximately 70% of the total blood volume which acts as a physiological reservoir (‘unstressed’ volume). Under conditions where circulatory demands increase, increased sympathetic tone will cause contraction of this reservoir. The resultant autotransfusion (‘stressed’ volume) may increase venous return by approximately 30% and subsequently cardiac output.2,3
Both the arterial and venous systems are integrated under complex neurohormonal influences. These include the adrenergic, renin–angiotensin–aldosterone, vasopressinergic and glucocorticoid systems in addition to local mediators such as nitric oxide, endothelin, endorphins and the eicosanoids.4
PATHOPHYSIOLOGY
HEART RATE FAILURE
Profound bradycardia will reduce both cardiac output and mean arterial pressure if sympathetic tone is compromised. Inotropes will increase both rate and speed of conduction, in addition to augmenting peripheral venous return, thereby restoring cardiac output and mean arterial pressure.
MYOCARDIAL FAILURE
Myocardial or ‘pump’ failure may be divided into disorders of systolic ejection (systolic dysfunction) and diastolic filling (diastolic dysfunction).
Diastolic dysfunction is characterised by reduced ventricular compliance or increased resistance to ventricular filling during diastole. It may be due to mechanical factors such as structural abnormalities of the ventricle (e.g. restrictive cardiomyopathy) or to impaired diastolic relaxation that occurs with myocardial ischaemia or severe sepsis. This results in elevated end-diastolic pressure and pulmonary venous congestion. Episodic or ‘flash’ pulmonary oedema is a common clinical sign of diastolic dysfunction.5 Tachycardias that shorten diastolic time may exacerbate diastolic failure. Diastolic dysfunction frequently accompanies systolic failure, in both acute and chronic cardiac failure, particularly in elderly patients.
VASOREGULATORY FAILURE
These syndromes are characterised by reduced responsiveness of the peripheral circulation to endogenous or exogenous sympathetic stimulation. This results in pooling in the venous circulation due to the inability to provide a ‘stressed’ volume.4
Management of these conditions has traditionally focused on the arterial circulation with attempts to increase systemic vascular resistance, often regarded inaccurately as treatment of ‘afterload failure’. This is a misnomer as the problem is predominantly impaired venous return, compounded to a lesser extent by pathological arteriolar vasodilatation.6 Clearly, the effects of vasoregulatory failure will be exacerbated by concomitant hypovolaemia. Fluid loading to restore effective intravascular volume is essential.
CLASSIFICATION
The common ultimate cellular mechanism of action of these agents involves an influence on the release, utilisation or sequestration of intracellular calcium (Figure 82.2). These agents are divided into two main groups based on whether or not their actions depend upon increases in intracellular cyclic adenosine 3,5-monophosphate (cAMP) and are outlined in Table 82.2.
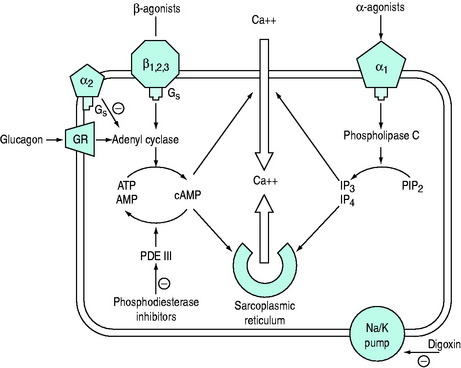
Table 82.2 Classification of inotropes
| cAMP dependent | cAMP independent |
|---|---|
| Catecholamines (β-adrenergic agonists) | Catecholamines (α-adrenergic agonists) |
| Adrenaline | Adrenaline |
| Noradrenaline | Noradrenaline |
| Dopamine | Dopamine |
| Dobutamine | Digoxin |
| Dopexamine | Calcium salts |
| Isoprenaline | Thyroid hormone |
| Phosphodiesterase inhibitors | |
| Amrinone | |
| Milrinone | |
| Enoximone | |
| Levosimendan | |
| Calcium sensitisers | |
| Levosimendan | |
| Glucagon |
CATECHOLAMINES
RECEPTOR BIOLOGY
PHYSIOLOGICAL
Agonists bind to populations of adrenergic receptors, largely divided into α and β subgroups. Further subgroups of α- (α1A, α1B, α2A, α2B, α2C) and β-receptors (β1, β2, β3) have been identified.7
PATHOPHYSIOLOGICAL
The activity and function of this system is dynamic and may be markedly influenced by pathological states. This mayresult in qualitative changes in the agonist–receptor–effector organ relationship (desensitisation) where receptors no longer respond to physiological or pharmacological sympathetic stimulation to the same extent. Quantitative changes such as reduced receptor density, receptor sequestration and enzymatic uncoupling (downregulation) may also result in impaired responses.8
BIOSYNTHESIS
The biosynthesis and chemical structures of the naturally occurring catecholamines are shown in Figure 82.3a.
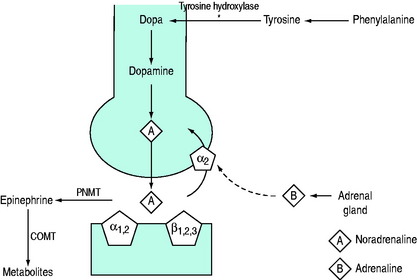
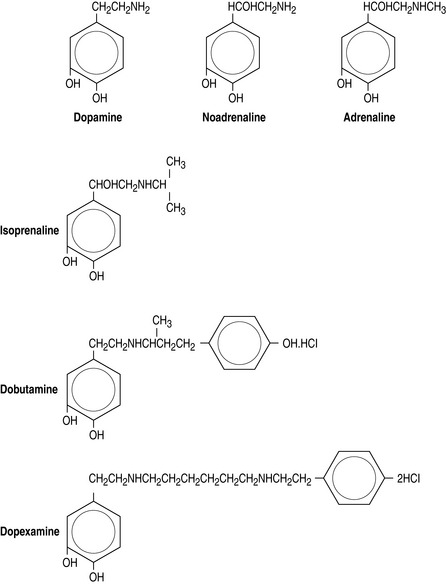
Figure 82.3 (a) Biosynthesis of catecholamines in sympathetic terminals. *Rate-limiting step by tyrosine hydroxylase. COMT, catechol-O-methyl transferase; PNMT, phenylethanolamine-N-methyltransferase. (b) Chemical structure of endogenous and synthetic catecholamines.
The synthetic catecholamines are derivatives of dopamine (Figure 82.3b). These agents are characterised by increased length of the carbon chain, which confers affinity for β-receptors. Dobutamine is a synthetic derivative of isoprenaline. These agents have relatively little affinity for α-receptors due to the configuration of the terminal amine, which differs from the endogenous catecholamines.
SYSTEMIC EFFECTS
The synthetic catecholamines are all predominantly β-agonists.
CARDIOVASCULAR
The cardiovascular effects of the catecholamines under physiological conditions are shown in Table 82.3.
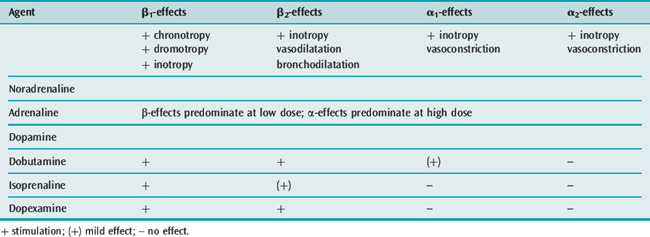
In the failing myocardium, particularly in patients with cardiac failure following cardiopulmonary bypass or septic shock, endogenous stores of noradrenaline are markedly reduced.9 Furthermore, there may be significant desensitisation and downregulation of cardiac β-receptors. In these situations, α1– and α2-receptors have an important role in maintaining inotropy and peripheral vasoresponsiveness.10 This may be expressed clinically as ‘tolerance’ or tachyphylaxis to catecholamines, particularly with predominantly β-agonists such as dobutamine. This phenomenon may explain the requirement for high doses of catecholamines in refractory shock states. Consequently, the role of β-agonists in patients with severe myocardial failure has been questioned.
Catecholamines have a significant effect on the venous circulation. These drugs primarily restore or maintain ‘stressed volumes’ of the capacitance vessels under pathological conditions, thereby maintaining or increasing cardiac output and mean arterial pressure. This is important in ‘vasoplegic’ states such as septic shock.11
In clinically used doses, intravenously administered catecholamines have minimal direct vasoconstrictive effects on conducting arterial vessels. Consequently, derived indices such as systemic vascular resistance do not reliably reflect the effect of catecholamines on the peripheral vasculature.



Full access? Get Clinical Tree


