endothelial and mesothelial cells of blood vessels. Cell death ensues, after which the rickettsia contiguously spread and infect adjacent cells. The infection stimulates a severe inflammatory response. This widespread, systemic vasculitis is directly responsible for almost all of the symptoms of RMSF.
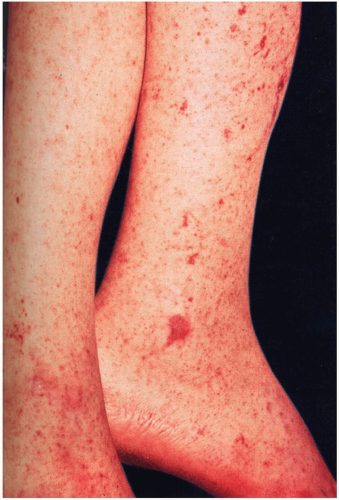 FIGURE 20-1 Rocky Mountain spotted fever. There is a generalized petechial eruption that involves the entire cutaneous surface, including the palms and soles. (From Habif, with permission.) |
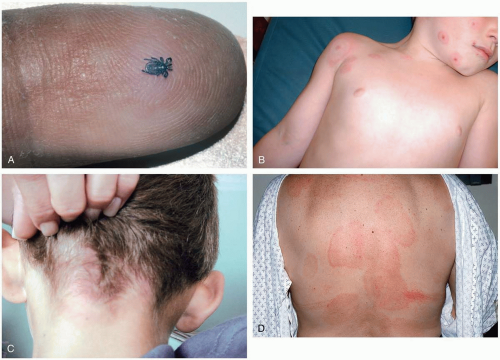 FIGURE 20-2 A: Ixodes tick. An adult tick is the size of the head of a match. (From Goodheart, with permission.) B: Multiple target erythema migrans lesions. (Courtesy of Alfredo Sabbaj, MD.) C: Target lesion of Lyme disease. (Courtesy of Christy Salvaggio, MD.) D: Erythema migrans lesion of Lyme disease. (From Goodheart, with permission.) |
spirochetes disseminate to various tissues; virulence factors influence which tissues are affected. B. burgdorferi remains in a constant state of antigenic flux during its life in the mammalian host, continuously triggering an inflammatory response, which may vary in severity. Chronic infection in susceptible individuals may result from this inflammatory response.3
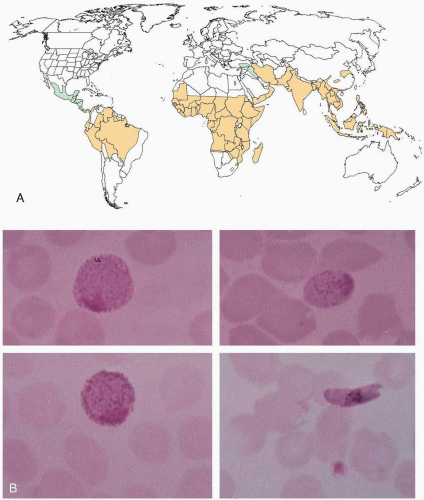 FIGURE 20-3 A: World distribution of malaria. Black areas indicate distribution of chloroquine-susceptible Plasmodium falciparum malaria, and gray areas indicate distribution of chloroquine-resistant Plasmodium falciparum malaria. (Modified with permission from Barat LM, Bloland PB. Drug resistance among malaria and other parasites. Infect Dis Clin North Am 1997;11:969-987.) B: Macrogametocytes of Plasmodium vivax (top left), Plasmodium malaria (top right), Plasmodium ovale (bottom left), and Plasmodium falciparum (bottom right). (From Smith JW. Atlas of diagnostic medical parasitology: blood and tissue parasites. Chicago: American Society of Clinical Pathologists, 1976.) |
TABLE 20-3 Selected Clinical Characteristics of Four Types of Malaria | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
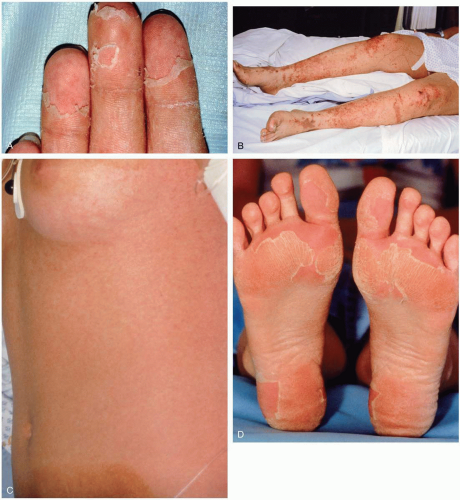 FIGURE 20-4 A: Desquamation of the palmar surface of the fingers. B: Petechiae and ecchymoses of the lower extremities. C: Petechiae of the trunk. D: Desquamation of the soles. (A–D, Courtesy of B. Zane Horowitz, MD.) |
TABLE 20-4 Case-Defining Parameters for Toxic Shock Syndrome (TSS) | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||
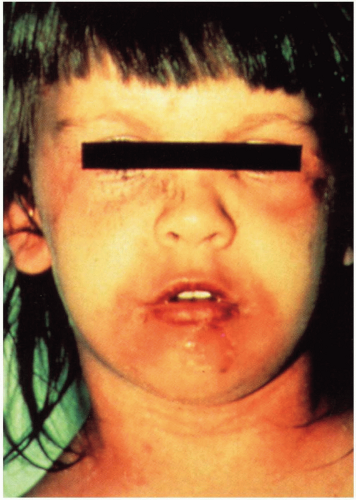 FIGURE 20-5 Staphylococcal scalded skin syndrome. Erythema is prominent on the neck and around the eyes and mouth. Crusting is also apparent in the periorificial areas. Over the chin, a bulla has ruptured, leaving a moist erosion. (From Mandell, Essential Atlas of Infectious Diseases, with permission.) |
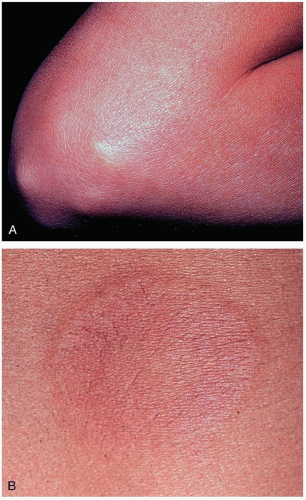 FIGURE 20-6 A: Subcutaneous nodules are generally associated with severe carditis. They are painless, firm, movable, measure approximately 0.5 to 2 cm, and usually are located over extensor surfaces of the joints, particularly knees, wrists, and elbows. B: Closer view of erythema marginatum in the same patient. (A and B, From Binotto MA, Guilherme L, Tanaka AC. Rheumatic fever. Images Paediatr Cardiol 2002;11:12-25.) |
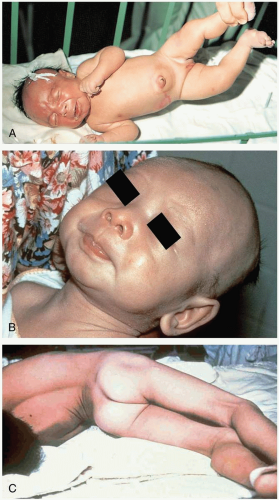 FIGURE 20-7 A: Neonatal tetanus at 6 days. The umbilical stump was treated with ashes. Note risus sardonicus and opisthotonus. (From Ostler, with permission.) B: “Lockjaw.” (Courtesy of the World Health Organization.) C: Opisthotonus. (Courtesy of the Centers for Disease Control and Prevention.) |
TABLE 20-8 Differential Diagnosis of Botulism | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 20-9 Drugs and Toxicants Associated with Hepatitis | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
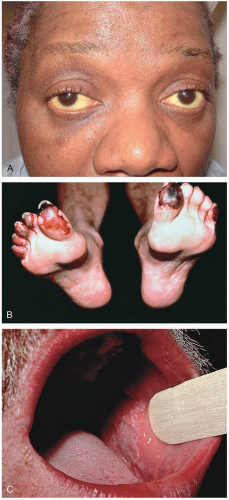 FIGURE 20-9 A: Icteric sclera in a patient with hepatitis C. (Courtesy of Mark Silverberg, MD.) B: Cryoglobulinemia produces acral vasculitic infarcts. Hepatitis C is a leading cause. (From Yamada, with permission.) C: Lichen planus produces lacy mucosal plaques and pruritic papules on the skin. Patients should be evaluated for hepatitis C infection. (From Yamada, with permission.) |
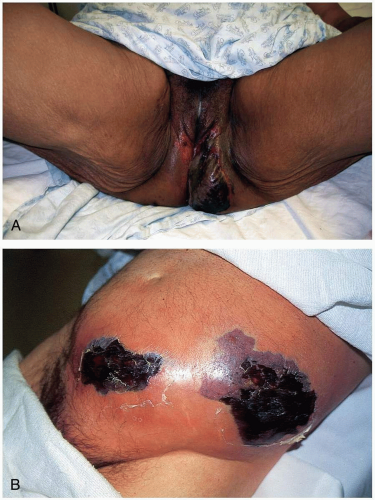 FIGURE 20-10 A: Fournier’s gangrene. (Courtesy of Mark Silverberg, MD.) B: Necrotizing fasciitis of the anterior abdominal wall. (From Isaacs L. Necrotizing fasciitis: diagnosis and treatment. Emerg Med News 2002;24[8]:4, with permission.) |
Related posts:







Full access? Get Clinical Tree


