Chapter 91 Infection and Host Response
Historical Perspectives
Traditionally, the immune system has been divided into innate and adaptive components. Clonal expansion of lymphocytes in response to infection is absolutely critical to the development of the immune response. However, it takes 3 to 5 days for clonal expansion to produce sufficient numbers of “effector” cells. Clearly, this is more than enough time for a pathogen to damage the host. The innate immune system is fundamental in eliminating the infection, and if not, then controlling it, until the adaptive immune responses eliminate it. If the innate and adaptive immune responses are “adequate,” the infection remains localized. If not, then the systemic response to infection, or “sepsis” results (see Chapters 90 and 103). What has become increasingly clear is that the adaptive and innate immune systems can each affect the functioning of the other.1
In the past 25 years, new concepts that are fundamental to the care of critically ill patient have arisen directly from our understanding of the host’s response to infection. (1) Inflammatory reactions first characterized as a response to infection are the foundation of a number of other pathogenic mechanisms, such as ischemia/reperfusion injury, direct trauma, drug-induced injury, inhalational injury, and multiple-system organ dysfunction. (2) The response to infection may lead to further injury of the host. Alternatively, an exuberant antiinflammatory response may likewise be deleterious, resulting in immune suppression and fibrosis (see Chapter 104). (3) There is a direct interaction between the neuroendocrine axis and inflammation (see Chapter 102). (4) There is a fundamental interrelationship among endothelium, inflammation, and coagulation, and efforts to intervene in one will likely have effects in others (see Chapter 101).
Innate Immune Versus Adaptive Immune Response
The innate immune system is phylogenetically ancient. Innate immune recognition is genetically predetermined (i.e., by germline-encoded receptors). Thus these receptors have evolved by natural selection, and have defined specificities for infectious organisms. In contrast, in the adaptive immune system, the T-cell and B-cell receptors are somatically generated in a way that gives each lymphocyte a unique structural receptor. Because the T- and B-cell receptors are not encoded in a germline, they are not predetermined to recognize any particular antigen. No matter how useful these “receptors” become, they cannot be passed down to the next generation. Although there are potentially a large number of variants of germline-encoded receptors (perhaps in the hundreds), there are 1014 and 1018 different somatically generated immunoglobulin receptors and T-cell receptors, respectively.1 Microbes are heterogenous and can mutate at very high rates. This can be handled appropriately by the adaptive immune system. This heterogeneity, however, represents more of a challenge for the innate immune system. As such, a different strategy, phylogenetically older than the adaptive system, evolved. The innate immune system has developed receptors that recognize “pathogen-associated molecular patterns” (PAMPs). PAMPs are highly conserved structures present in a large group of microorganisms. The common features of PAMPs are (1) PAMPs are only produced by microbial pathogens, not hosts; (2) structures recognized by the innate immune system are usually essential for survival or pathogenicity of the microorganism; and (3) PAMPs are usually invariant structures shared by an entire class of pathogen. The best known examples of PAMPs are bacterial lipopolysaccharide (LPS), peptidoglycan, lipoteichoic acid (LTA), mannans, bacterial DNA, double-stranded RNA, and glucans.2
Pattern Recognition Receptors
Toll-Like Receptors
At present there are nine human TLR receptors.8 The TLRs recognize molecules common to pathogens ranging from protozoa to bacteria to fungi and to viruses. The TLRs that recognize bacterial cell wall components (TLR1, -2, -4, and -6) are located on the cell surface, forming homodimers and heterodimers. Once ligated, they then translocate to the endosome for signaling (see Chapter 103).9 TLR3, TLR7, and TLR9 reside in the endosome, recognize nucleic acids produced by viruses and bacteria, and thus are available for activity against intracellular viruses.10
There are several other classes of PRRs in addition to TLRs: RIG-I–like receptors (RLRs), Nod-like receptors (NLRs), and C-type lectin receptors (CLRs) (previously described). RLRs, an intracellular receptor, recognize single- and double-stranded viral RNA and transmit their signal through a common adaptor protein, interferon promoter stimulator-1 (IPS-1) and NF-κB, to induce type I interferon production and antiviral responses.10 Double-stranded RNA-dependent protein kinase (PKR) is a unique PRR in that it can turn off protein translation directly; its effect does not seem to be modulated by downstream effectors.14 Nod1 and Nod2 are NLRs that recognize PAMPs derived from the bacterial cell wall. Nod1 and Nod2 are localized to the cytoplasm and can elicit TLR-independent antibacterial responses. Other NLRs, such as NALP1, NALP3, IPAF, and NAIP5, are components of a molecular complex called the inflammasome. The inflammasome complex comprises one or some of the NLR proteins and caspase-1. Caspase is activated in this complex and cleaves critical inflammatory molecules such as pro-IL-1β and pro-IL-18 to produce mature proteins.10 The NALP3 inflammasome is activated by stimuli such as uric acid crystal, silica, and asbestos; thus NALP3 appears to be a receptor for danger-associated molecular patterns. Infections with the malaria parasite or the fungus Candida albicans were also reported to activate the NALP3 inflammasome.
What follows is a focus on the pathogen ligands of the PRRs, in particular TLR4. It had been recognized for a number of years that CD14, present on monocytes and blood-derived macrophages, and lipopolysaccharide binding protein (LBP), found in plasma, were critical for an LPS response. However, CD14 is a glycosylphosphatidylinositol-anchored protein lacking transmembrane and intracellular domains and would be unable to signal intracellular processes. CD14 is also present to a limited extent on neutrophils.15 In addition, a soluble form of CD14 could substitute for membrane-bound form of CD14 in LPS-mediated signaling. Thus it was clear that at least one other molecule present on the cell surface would be needed to elicit an LPS-dependent response. TLR4 knockout mice and human TLR4 mutations conferred such a role for this receptor for LPS-induced responses in humans and mice. TLR4 requires an additional molecule, MD-2, that is part of the complex on the cell surface. LPB is an acute phase reactant.2,16 It catalyzes the transfer of LPS to CD14, which is then able to interact with TLR-4 and MD-2, resulting in intracellular signaling through the Toll/IL-1 receptor homologous region (TIR) adaptor molecules.9 Downstream signaling events result in release of transcriptional activating factors including NF-κB, that guide gene transcription of many genes, including TNF and interferon-β (IFN-β), a type I interferon. TNF is discussed at greater length later in this chapter; however, it is a central mediator in innate immune response pathway. IFN-β is critical in adaptive and antiviral immunity (see Chapter 103 for a description of other TLRs).
Soluble Components of Immunity
Complement System
The complement system is critically positioned to participate in both the innate and adaptive immune response.26 However, it is also critical for the disposal of immune complexes from tissues and clearance of apoptotic cells.4 The three pathways in which complement may become activated are the classic, alternative, and mannose-binding lectin pathway. Although complement activation initiates differently in each, all three converge at the cleavage of C3 (Figure 91-1). The classic pathway is initiated by the binding of the C1 complex (consisting of C1q, C1r, and C1s) to antibodies bound to antigen on the cell wall. The mannose-binding lectin pathway is initiated by binding of the complex of mannose-binding lectin and the mannose-binding lectin-associate proteases 1 and 2 (MASP1 and MASP2) to arrays of mannose groups on the surface of the bacterial cell wall. Both MASP2 and C1q then function similarly in forming the C4bC2a complex, representing the convertase for C3. Bacterial products, LPS, yeast cell wall particles, and aggregated antibody, including immunoglobulin (Ig)A and IgE, are all capable of activating the alternative pathway. The alternative pathway is initiated via low-grade cleavage of C3 in plasma to C3b. C3b binds to the hydroxyl groups on cell surface carbohydrates. C3b binds factor B to form a C3bB complex, which is activated by factor D, forming C3bBb and stabilized with properdin. C3bBb then functions as an alternative convertase for C3, that cleaves many molecules of C3 to C3b. C3b binds to hydroxyl groups on the microorganism around the area of complement activation. C3a, an anaphylatoxin, is released by the C3 convertase. C3b can also bind to the C3 convertase to form C5 convertase. The C5 convertase releases the anaphylatoxin C5a and initiates the formation of the membrane attack complex (MAC), C5b6789.4 The membrane attack complex created inserts into the cell membrane, creates large pores, and leads to osmotic lysis of the target.
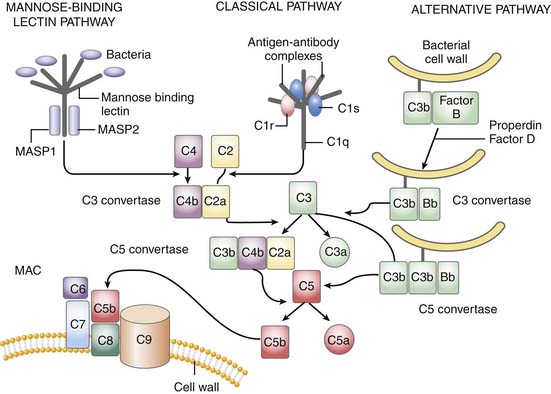
Thus the complement system amplifies the initial response to the organism. In addition to lysing the target organism, opsonization with complement fragments C3b/C4b and C3bi (fragment C3b) occurs, resulting in phagocytosis by neutrophils, monocytes, and macrophages. Binding occurs through specific complement receptors, CR1 for C3b/C4b (CD35) and CR3 for C3bi (CD11b/CD18, also known as Mac-1 and αMβ2). The anaphylatoxins C3a, C4a, and C5a produced are low molecular weight, biologically active peptides defined by their actions on small blood vessels, smooth muscle, mast cells, and peripheral blood leukocytes. Blood vessels, smooth muscle cells, basophils, and mast cells respond to all three anaphylatoxins. Neutrophils, monocytes, and macrophages respond to C5a. The anaphylatoxins promote edema and increase vascular permeability through the release of histamine from mast cells and through the local production of vasodilatory prostaglandins such as prostaglandin E2 (PGE2) and edemogenic leukotrienes (LT) C4, D4, and E4. C5a is a powerful activator of granulocyte function including chemotaxis, degranulation, and increased oxidative metabolism. C5a actions occur through its seven-transmembrane–spanning, G protein–coupled receptor, C5aR.27 In animal models, C5a/C5aR is critical to the development of sepsis and multiple organ failure, and potentiates many early response cytokines and coagulation. C5aR is present not only on leukocytes, but also other tissue, including the brain, kidney, gastrointestinal tract.27,28
Complement activation also occurs after oxidative stress such as occurs with ischemia/reperfusion. Complement activation is an early event after injury and the inhibition of complement activation or its components offer tissue protection after reperfusion.29 Finally, complement proteins transduce various cell signals. Complement can activate B and T cells. It can regulate apoptosis of various cell types (see Chapter 100).20,26
Immunoglobulin
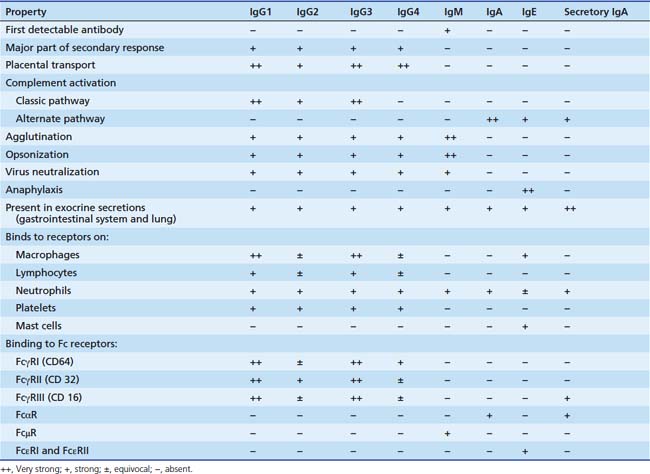
The different immunoglobulins (Igs) secreted by B cells (IgG, IgA, IgM, IgD, and IgE) are known as isotopes. IgD plays little role, if any, in containment of microorganisms. Ig isotypes can be divided into subclasses, variants that show slight structural differences but are sufficiently alike structurally to be essentially identical to other members of the isotype class. There are four IgG subclasses: IgG1, IgG2, IgG3, and IgG4. As seen in Table 91-1, isotype subclasses have specialized roles in the immune response. For example, IgG2 has a major role in the formation of carbohydrate antibodies, but has poor complement fixation characteristics. The antibody system consists of Ig present in serum, protecting the blood and tissue spaces, and that present in the secretory system, lining the gastrointestinal and respiratory tracts and present in tears. The serum component is mostly IgG (85%), with lesser amounts of IgA and IgM. The secretory system consists mostly of secretory IgA (85%), which is structurally different than serum IgA, and lesser amounts of IgG and IgM. All Igs have a basic four-chain structure composed of an identical pair of heavy (H) and light (L) chains. The H-L pairs are held together by interchain disulfide bonds and noncovalent forces. The binding site for antigen is formed by one H and one L chain. IgM is a polymer of five four-chain units, and secretory IgA is a dimer. Polymeric forms (Figure 91-2) possess a J chain that is synthesized with the H and L chains and serves to stabilize the sulfhydryl groups during polymerization. At regular intervals along the peptide chain a disulfide bond forms an intrachain loop, known as the Ig domain. This motif is repeated among immunoglobulins, T-cell receptors, adhesion proteins, and histocompatibility antigens. Proteins with these Ig domains share close homology structurally and functionally and suggest a common evolutionary origin. They are termed members of the Ig superfamily (IgSF), and all are involved in cell interaction processes associated with recognition.
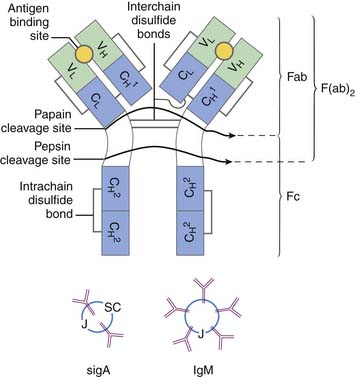
The Ig molecule is divided into regions, the amino acid sequence of which is similar, such as regions needed for complement fixation or attachment to receptors on leukocytes. However, other regions are highly variable; those that bind to antigen have the highest divergency and are collectively known as the hypervariable region. Thus each Ig chain can be divided into constant and variable regions. The H chain has three constant regions and one variable region; the L chain has one variable region and one constant region. The hypervariable region of the L and H chain is tightly apposed and forms the combining site for antigen (see Figure 91-2). The digestion of Ig with papain and pepsin generates fragments with varying biologic capability. The Fc region of the Ig molecule accounts for its isotypic biologic capability. This is the region critical for complement fixation and recognition by Fc receptors on the leukocytes. The Fab region provides for specific unique antigen-antibody interactions. The F(ab′)2 fragment is formed with pepsin cleavage; the affinity for antigen is twice as great as Fab alone.
Contact Activation System
There are four major plasma protein systems that contribute to the host’s defense and participate in the development of inflammatory tissue injury: the complement system (previously discussed), the contact activation system (also known as Hageman factor or intrinsic coagulation system), the extrinsic coagulation cascade, and the fibrinolytic system (see Chapter 80). The contact activation system is critical to host defense and control of local blood flow at sites of injury. Hageman factor (Factor XII) is activated spontaneously (XIIa) on contact with negatively charged surfaces, such as lipid A of LPS and vascular basement membranes (Figure 91-3). High-molecular-weight kininogen (HMWK), prekallikrein, and factor XI circulate in the plasma as complexes. Factor XIIa will activate factor XI and cleave prekallikrein to kallikrein. Kallikrein will then cleave HMWK to bradykinin. The kallikrein-kinin system also encompasses the tissue (or glandular) kallikrein-kinin system (see Figure 91-3). Tissue kallikrein is immunologically distinct from plasma kallikrein and present throughout the body as an inactive “pro-” substance. In the presence of intracellular enzymes, plasmin, or plasma kallikrein, tissue kallikrein is produced, secreted, and active in the tissue where it is made. Tissue kallikrein can then cleave HMWK to bradykinin directly, or it can cleave low-molecular-weight kininogen (LMWK) or tissue kininogen (T-kininogen) to kallidin that is then directly converted to bradykinin. In the plasma, 80% of kininogen is low molecular weight.
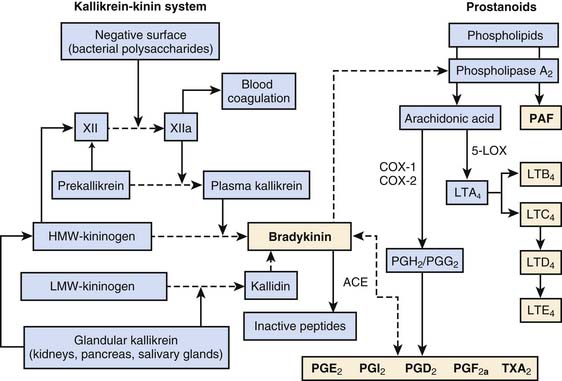
Bradykinin is an exceedingly potent vasoactive peptide.31 It can cause venous dilation, increased vascular permeability, hypotension, bronchoconstriction, and activation of phospholipase A2. Phospholipase A2 releases arachidonic acid from cell membrane and initiates the production of both proinflammatory and antiinflammatory phospholipids-derived products (see subsequent section and Figure 91-3). Bradykinin is metabolized by angiotensin-converting enzyme (ACE) to inactive peptides. Bradykinin, along with prostanoids, stimulates the pain response through polymodal receptors and C-fibers (capsaicin sensitive).32 Bradykinin has been shown to be responsible for the four signs of inflammation: heat, redness, swelling, and pain. The bradykinin effect is enhanced by simultaneous production of prostanoids. Prostanoids, particularly along with the low pH of exudates, inhibit the activity of kininases such as ACE.






Full access? Get Clinical Tree


