Chapter 63
Idiopathic and Thrombotic Thrombocytopenias
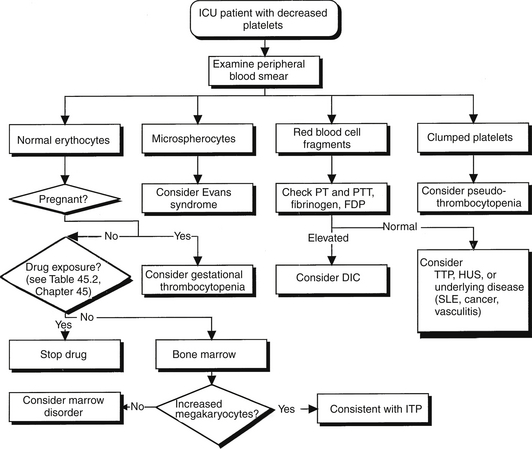
Moderate to severe thrombocytopenia is a major feature of several disorders that accompany or lead to stays in the intensive care unit (ICU). Immune (idiopathic) thrombocytopenic purpura (ITP) and thrombotic thrombocytopenic purpura (TTP) are two such conditions. Early and accurate diagnosis and rapid application of appropriate therapy are keys to the successful management of patients with these disorders. Accurate diagnosis requires differentiating the thrombocytopenia of ITP and TTP from each other as well as from other thrombocytopenic conditions. The diagnostic workup should begin with a carefully focused history and physical examination, followed by an examination of the peripheral blood smear (Figure 63.1). Blood products should not be given to a nonbleeding patient with thrombocytopenia until the smear is inspected, particularly to evaluate for the presence or absence of schistocytes and rule out the presence of TTP. Moreover, during the initial evaluation, patients should be safeguarded from iatrogenic bleeding complications resulting from a low platelet count (Box 63.1).
Immune Thrombocytopenic Purpura
Mechanism and Diagnosis
ITP remains a diagnosis of exclusion after other causes of thrombocytopenia are ruled out. Box 63.2 lists the differential diagnosis of thrombocytopenia without microangiopathy. Evans syndrome is also an antibody-mediated disease, with antibodies directed against both platelets and erythrocytes. The destruction of erythrocytes occurs with the same mechanism as does the destruction of platelets. Bone marrow aspiration is an appropriate test to establish the diagnosis in patients older than 60 or in those considering splenectomy.
BOX 63.2 Differential Diagnosis of Thrombocytopenia without Microangiopathy∗

∗Microangiopathy denotes the presence of schistocytes (fragmented red blood cells) on peripheral smear. ITP, immune thrombocytopenic purpura.
ITP has been associated with other conditions in up to 20% of cases. In this setting, it is often called secondary ITP. Causes of secondary ITP include collagen vascular diseases, lymphoproliferative disorders, infections, and other autoimmune syndromes. Thrombocytopenia is also a common manifestation of human immunodeficiency virus (HIV) disease, presumably caused by increased platelet destruction on an immune basis. The clinical features of HIV-associated ITP are similar to those of idiopathic ITP. However, ITP associated with HIV infection in patients with hemophilia is of particular concern because these patients are at much higher risk for catastrophic bleeding than are patients with primary ITP.
Treatment
Glucocorticoids have become the mainstay of treatment for ITP that is symptomatic or when the platelet count falls to < 30,000/μL. The typical daily dose of prednisone is 1 mg/kg. This inexpensive treatment elevates platelet counts in about two thirds of treated patients. Most patients exhibit a decline in platelet count when the prednisone dose is tapered and therefore require additional therapy. The anti-CD20 monoclonal antibody, rituximab, has shown efficacy in patients who relapse following glucocorticoid therapy. Rituximab is typically given by monthly intravenous infusions of 375 mg/m2 for four doses. Alternatively, anti-D immune globulin has shown efficacy in patients with ITP who are Rh positive and have an intact spleen. Splenectomy can produce sustained increases in platelet counts in more than two thirds of patients with ITP. Pneumococcal, meningococcal, and hemophilus vaccines should be given 2 weeks before splenectomy to reduce the risk of severe complications resulting from postsplenectomy pneumococcal bacteremias. Some patients have a relapse of ITP months or even years after splenectomy. This is attributable to the presence of an accessory spleen. These spleens occur in up to 20% of patients and are easily detected by radionuclide scanning. After the removal of accessory spleens, ITP resolves in many of these patients.


Full access? Get Clinical Tree


