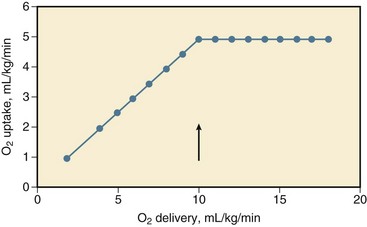
26 Although hypovolemic shock has been recognized for more than 100 years, Wiggers1 in 1940 first offered a definition of hypovolemic shock that has remained significant until now: “Shock is a syndrome resulting from depression of many functions, but in which the reduction of the effective circulating blood volume is of basic importance, and in which impairment of the circulation steadily progresses until it eventuates in a state of irreversible circulatory failure.” Today, hypovolemic shock can be defined as an acute disturbance in the circulation leading to an imbalance between oxygen supply and demand in the tissues, caused by a decrease in circulating blood volume, mostly caused by trauma and hemorrhage.2 An oxygen debt develops when uptake no longer matches the demand for oxygen and leads to cellular ischemia and ultimately cell death. The condition is life threatening and, if left untreated, becomes irreversible after a certain period. Rapid and adequate resuscitation is mandatory to save lives. Conversely, hypovolemic shock carries a relatively favorable prognosis, if rapidly and adequately recognized and treated. Hypovolemic shock can occur outside and inside the hospital, in trauma or surgery complicated by excessive loss of blood, but also in the course of burns, gastrointestinal hemorrhage, diarrhea, uncontrolled diabetes mellitus, addisonian crisis, and other conditions (Box 26.1). Some other types of shock, including septic, anaphylactoid, cardiogenic, and burn shock, may be accompanied by hypovolemia. The types of shock not primarily caused by hypovolemia are beyond the scope of this chapter. Because hypovolemia results in a decrease in preload of the heart and low filling pressures or volumes, the cardiac output decreases.3–9 After unloading of the baroreceptor and activation of the sympathetic nervous system, tachycardia ensues, although some patients may respond with transient sympathetic inhibition and vagal nerve–mediated bradycardia during a sudden, severe loss of circulating blood volume.3,10–16 Tachycardia partially compensates for the decrease in stroke volume. A moderate decrease in cardiac output can be recognized from a decline in pulse pressure, orthostatic hypotension, and fall in regional perfusion indices.8,17,18 Hypovolemia results in wider than usual swings in central venous pressure (CVP) and arterial blood pressure during the respiratory cycle of spontaneous and mechanical ventilation because of increased sensitivity of the underfilled heart in the ascending part of the cardiac function curve to fluctuations in venous return associated with varying intrathoracic pressure.19,20 Although activation of the sympathetic nervous system and resulting arterial vasoconstriction during a moderate decrease in cardiac output prevent a severe reduction in arterial blood pressure, a further decrease in cardiac output leads to hypotension and shock.8,10 Systemic vascular resistance increases early after development of hypovolemic shock but may decrease in the later stages of shock, and this may herald irreversibility and death. The increase in resistance (and heart rate) may be transiently attenuated after an imbalance between sympathetic and vagal activity, possibly associated with release of opioids within the central nervous system and into the systemic circulation.* Shock is characterized by an oxygen debt in the tissues.9,24–26 In the presence of sufficient oxygen, aerobic combustion of 1 mol of glucose yields 38 mol of energy-rich adenosine triphosphate (ATP), which can be hydrolyzed to provide energy for the vital and metabolic functions of the cell.27 In the absence of oxygen, glucose taken up by cells cannot be combusted because of insufficient uptake of pyruvate into the mitochondrial tricarboxylic acid cycle having a reduced turnover rate. Partly inactivated pyruvate dehydrogenase may play a role in the latter reductions. Pyruvate is converted into lactate, and the lactate-to-pyruvate ratio increases, concomitantly with a reduction in mitochondrial redox potential.24,27–29 Anaerobic glycolysis in the cytosol ultimately yields, per mol of glucose, 2 mol of ATP.27 Hydrolysis of ATP yields hydrogen ions (H+) that lead, when buffers are exhausted, to intracellular and ultimately to extracellular metabolic acidosis.30 These mechanisms form the basis of the so-called lactic acidosis during hypovolemic shock, whereby the lactate level in arterial blood is elevated above the normal 2 mmol/L associated with acidosis, and constitutes a useful measure of the oxygen debt in the tissues.26,27,31–34 Nevertheless, the energy deficit and lactate production in the cells in response to a lack of oxygen can be limited and organ function can be improved by supplying pyruvate and pyruvate dehydrogenase activators, such as dichloroacetate.27,29,35–37 Intracellular acidosis may otherwise protect ischemic cells from dying.36 The specificity of elevated lactate-to-pyruvate levels for an oxygen debt in the tissues has been doubted.27,38 Aerobic glycolysis is probably linked to the membrane Na+/K+-ATPase and stimulation of β2-receptors during sympathetic activation. Catecholamine (epinephrine) secretion may temporarily increase, rather than decrease, ATPase activity, and augment glycolysis and circulating lactate levels in tissues such as skeletal muscle, without a lack of oxygen and reduced ATP resources, during development and resuscitation from hypovolemic shock.38,39 Conversely, adrenergic antagonists may reduce lactic acidosis during hypovolemic shock.38 Epinephrine may increase glycogenolysis. Together, increased glycolytic fluxes independent of oxygen uptake may lead to equal elevations of pyruvate and lactate in the tissues, without the acidosis resulting from ATP hydrolysis with an oxygen debt.27 This situation may partly explain why the extent to which changes in the lactate level parallel changes in the anion gap or bicarbonate/base excess concentration during shock and resuscitation is controversial, and why elevated lactate levels sometimes may fail to predict an increase in oxygen uptake during an increase in oxygen delivery.40–42 This also may explain in part the discrepancies in the course of oxygen-related variables and lactate levels during catecholamine treatment of shock when attempting to boost oxygen delivery.42 The lactate level in blood is determined by production, distribution, and elimination.27 Produced lactic acid in the presence of oxygen may be converted via pyruvate to glucose or oxidized. Bicarbonate is then released. The liver plays a central role in this process, so that the elimination of lactate and clearance from plasma is impaired in case of liver ischemia or prior hepatic disease, even though renal uptake may increase.27,43,44 Nevertheless, changes in the lactate level in blood, rather than absolute values, mainly reflect changes in production and are a fair measure for the course of shock and the response to therapy, even in the presence of liver disease.27,45 Although not beyond doubt, the origin of lactate in hypovolemic shock can be skeletal muscle, lung, and gut, particularly if severe liver ischemia, hypoxia, and acidosis in shock attenuate the hepatic uptake of lactate delivered by the gut through the portal vein.27,43,44,46–48 The respiratory muscles also may contribute to lactic acidosis in a spontaneously breathing patient because, first, the respiratory muscles may demand a share of the cardiac output at the cost of other tissues, and, second, this share may be insufficient to meet oxygen demands of the diaphragm, which may be increased in view of hyperventilation.33,49–52 Notwithstanding the aforementioned limitations, an increase in the lactate level in blood and a decrease in the bicarbonate content/base excess or pH and an increase in the anion gap may be fair predictors of morbidity (multiple organ failure [MOF]) and mortality, whereas clearance of lactic acidosis usually indicates a better outcome. A decrease in the blood lactate level during resuscitation from hypovolemic shock is usually a favorable sign and associated with survival, whereas an increase in the lactate level and progressive acidosis usually are associated with morbidity and mortality, even though successful resuscitation may transiently increase the lactate level because of washout of lactate from ischemic tissues.* The mentioned variables may thus serve as guides for resuscitation. Because insufficient uptake of oxygen relative to demand in the tissues during shock is central, insight into the factors that determine oxygen uptake in shock is important.25,27 Oxygen delivery is determined by the cardiac output and the content of oxygen in arterial blood, that is, the arterial blood hemoglobin concentration and the saturation of hemoglobin with oxygen. The oxyhemoglobin dissociation curve determines the saturation of hemoglobin with oxygen for a given partial pressure of oxygen (PO2) in blood. During hypovolemic shock, a decrease in hemoglobin concentration, oxygen saturation, or both aggravates the effect of a decrease in cardiac output in compromising oxygen delivery to the tissues. Cardiac output is determined by preload, afterload, contractility, and heart rate.7 During a decrease in oxygen delivery with hypovolemic shock, the body maintains sufficient uptake of oxygen only if the extraction of oxygen increases, and the arteriovenous oxygen content gradient widens, resulting in a decrease in oxygen saturation of venous blood.† Associated with a decrease in oxygen delivery, tissue PO2 declines, and its heterogeneity increases, possibly indicating focal ischemia.‡ The decline in tissue PO2 may be even greater than the decrease in draining venous blood because of some increase in microvascular oxygen shunting at low blood flows.68,69 In animals, it has been shown that the increase in oxygen extraction to compensate for a decrease in oxygen delivery is maximum (but not 100%) if oxygen delivery decreases to less than 8 to 15 mL/kg per minute, that is, the critical oxygen delivery (Fig. 26.1).§ Although the critical oxygen delivery may vary widely among studies, following differences in species, basal oxygen needs, and methods to decrease oxygen delivery, data obtained in patients suggest that the critical oxygen delivery in humans may also amount to approximately 8 mL/kg per minute.58,64 During a decrease in oxygen delivery below this critical value in hypovolemic shock, oxygen uptake decreases to less than tissue demand, cellular ischemia ensues, and the body must rely on anaerobic metabolism to meet energy requirements.¶ Blood lactic acidosis, lacticacidemia, results. Conversely, oxygen uptake is supply-dependent if oxygen delivery is lower than the critical value and blood lactate levels are elevated, whereas oxygen uptake may not be supply-dependent if the lactate level in blood is normal. The critical oxygen delivery is a function of the body oxygen needs and the capability of the body to extract oxygen during a decline in delivery. The body oxygen needs may increase during hypovolemic shock, as a consequence of increased respiratory muscle activity and increased levels of catecholamines in the blood after activation of the sympathetic nervous system, but downregulation of the metabolic stimulant effect of catecholamines has been described.† The critical extraction of oxygen is a function of the adaptation of regional blood flow to tissue needs, the number of perfused capillaries and of diffusion distances, and the exchange surface area for oxygen.63,75 During a reduction in oxygen delivery, however, oxygen uptake is limited by convective transport of oxygen to the tissues, rather than by diffusion of oxygen to respirating mitochondria.76 In experimental animals, a change in hemoglobin affinity for oxygen, by altering the storage duration of reinfused blood, hardly changes the critical oxygen extraction, but changes in acid-base status that affect the position of the oxyhemoglobin dissociation curve may have some effect on the oxygen extraction capabilities of the body.60,76 Acid infusion may increase slightly, and base infusion may reduce oxygen extraction during supply-limited oxygen uptake.76 Nevertheless, hypercapnia may decrease critical oxygen extraction and increase critical oxygen delivery because of blood flow redistribution.77 A leftward shift of the oxyhemoglobin dissociation curve may impair maximum oxygen extraction during a reduction in oxygen delivery and may increase mortality rate in experimental animals with hypovolemic shock.60 Although the oxyhemoglobin dissociation curve may shift to the left in critically ill patients, for example, after transfusion of old, stored blood,78 the effect on oxygen uptake is unclear. The effect of changes in body temperature is twofold: Changes are accompanied by changes in total body oxygen needs and by changes in critical oxygen extraction, probably by a vascular tone–associated altered distribution of blood flow.61 Hyperthermia increases critical oxygen delivery in hemorrhaged dogs, primarily through an increase in body oxygen needs and despite an increase in critical oxygen extraction, whereas hypothermia, which may be more common in traumatized or hemorrhaged patients, may decrease the critical oxygen delivery.61 Finally, blood viscosity may influence the extent to which a decrease in circulating blood volume affects oxygen uptake by the tissues. Experimental data suggest, however, that prior anemia does not ameliorate the decrease in oxygen uptake during a decrease in oxygen delivery with hypovolemic shock, indicating that the convective transport of oxygen is the major determinant of oxygen uptake when delivery is impaired, even though prior hemodilution may increase oxygen extraction capabilities and decrease critical oxygen delivery.6,70 Taken together, these factors may influence the extent to which oxygen uptake decreases during reduced delivery and how far oxygen delivery should be enhanced during resuscitation from hypovolemic shock. The critical oxygen delivery varies among tissues. The oxygen needs of the kidney may decline during a decrease in renal oxygen delivery because a decrease in renal perfusion may lead to a reduction in glomerular filtration and to a reduction in energy-consuming tubular resorption.63 In contrast, during progressive hypovolemia, the gut may experience supply dependency of oxygen uptake earlier than nongut tissue, partly because of a higher critical oxygen delivery (higher needs and less extraction of oxygen) and partly because of redistribution of blood flow away from the gut mucosa after more intense vasoconstriction in gut than in nongut tissue.* Clinically, this may result in nonocclusive bowel ischemia. Respiratory muscles may also have a higher critical oxygen delivery than the body as a whole during progressive hemorrhage. Concomitant with an increased arteriovenous oxygen extraction during a decrease in oxygen delivery, the arteriovenous gradient of the carbon dioxide (CO2) content widens.8,52,81 The latter is associated with an increase in tissue and venous partial pressure of carbon dioxide (PCO2) relative to arterial PCO2 and a decrease in venous pH exceeding the decrease in pH in arterial blood.44,52,68 This widening of gradient is caused by the Fick principle and a greater decline in cardiac output than in oxygen uptake and CO2 production in the tissues because of inhibited oxidative metabolism. Nevertheless, the oxygen uptake usually decreases more than CO2 production, leading to an increase in respiratory quotient.49,65 This increase is likely to be caused by buffering of lactic acid by bicarbonate in the tissues and effluent blood, a shift toward glucose instead of fat use for residual oxidation in ischemic tissues, or a combination of both. The end-tidal expiratory CO2 fraction decreases in association with a reduction in oxygen uptake and CO2 production for a given ventilation.65 Conversely, a decrease in arterial PCO2 during a decline in CO2 production versus ventilation may be attenuated by an increase in dead-space ventilation resulting from a decrease in pulmonary blood flow/ventilation ratio.49 An increase in deadspace ventilation leads to widening of the gap between the arterial and expiratory PCO2.49 It has been suggested that the severity and duration of the oxygen debt accumulated during hypovolemic shock is a major determinant of survival in animals3,6 and in patients with trauma/hemorrhage and after major surgery.26,42,82 After trauma and hemorrhage, the defect in circulating blood volume and tissue oxygenation may be greater in patients who develop acute respiratory distress syndrome (ARDS) and MOF than in patients without these complications.* In patients undergoing major surgery, the oxygen debt during and after surgery may relate directly to the development of postoperative organ damage (i.e., MOF) and demise.73,82 Conversely, a high oxygen delivery and uptake during resuscitation may be associated with survival, whereas values that may be too low for elevated tissue demands are believed to contribute to ultimate demise, at least in animals with hypovolemic shock and critically ill patients after trauma or major surgery.† An increase of oxygen delivery and oxygen uptake to supranormal values has been suggested to improve survival further, although the latter debate has not been settled yet.‡ Extensive ischemic mitochondrial damage may limit an increase in oxygen consumption during resuscitation and reperfusion. During loss of blood volume, various mechanisms come into play that may counteract the resultant decrease in cardiac output and tissue oxygenation. First, a decrease in cardiac output during hypovolemic shock results in a redistribution of peripheral blood flow.§ This redistribution is partly the result of regional autoregulation to maintain blood flow, in which endothelial cells and production of endogenous partly gaseous vasodilators, including endothelial nitric oxide synthase–derived nitric oxide (NO), heme oxygenase–derived carbon monoxide, hydrogen sulfide, and metabolic by-products in the tissues including CO2, potassium, and adenosine, may play a central role.86–94 Endothelium-derived NO relaxes underlying smooth muscle in the vessel wall, via stimulation of guanylate cyclase and cyclic guanosine monophosphate (cGMP), which can be inhibited by methylene blue.87,88,95,96 Carbon monoxide also acts via cGMP.91 Some authors describe that inhibition of endothelial NO synthase ameliorates early hypotension and even the mortality risk during bleeding.92 When NO is released, the reactivity to endogenous and exogenous vasoconstrictors may be diminished, even early in hypovolemic shock.92,97 Other authors describe endothelial injury and dysfunction in various organs with diminished endothelium and NO-dependent vasorelaxation, which could be overcome by L-arginine and other NO donors, including ATP-MgCl2, pentoxifylline, or heparin, so that blockade of endothelial NO synthase–derived NO may be detrimental.87–89,97,98 The opposing vasoconstricting factors include catecholamines, liberated by the activated sympathetic nervous system and the adrenal medulla; direct sympathetic stimulation of the vessel wall; angiotensin II, liberated through an activated renin-angiotensin-aldosterone system; and vasopressin, released by the pituitary in hypovolemic shock.* Endothelin is an endothelium-derived potent vasoconstrictor, released on catecholamine stimulation or hypoxia, and its release may contribute to vasoconstriction, particularly in hepatic and renal vascular beds.100,101 Finally, a decrease in cardiac filling may reduce cardiac secretion of atrial natriuretic peptides, reducing the vasodilating and diuretic effect of these factors.102 Levels may also increase as a consequence of diminished renal clearance.103,104 Depending on the degree that the mechanisms are operative, the general result of the interplay is that blood flow to intestines, skeletal muscle, and skin is diverted toward vitally more important organs, such as heart and brain, so that the increase of overall peripheral resistance during hypovolemic shock is distributed differently among various organs, with greater increases in gut, skeletal muscle, and skin than in heart and brain.† The kidney also is a target for hypovolemic shock; renal perfusion may be maintained during mild hypotension after hypovolemia, but it rapidly decreases if severe hypotension supervenes, and the decrease may exceed that in other organs.‡ In hypovolemic human volunteers, this redistribution of blood flow accords with the patterns described.74 The redistribution of blood flow results in a greater share of oxygen delivery going to organs with high metabolic demand, such as heart and brain, than tissues with less metabolic demands, including skin, skeletal muscle, kidney, gut, and pancreas.§ The redistribution is probably necessary to optimize the uptake of delivered oxygen to the tissues and partly accounts for the increase in oxygen extraction during a decrease in oxygen delivery.63,75 In dogs, the ability of the body to extract oxygen diminishes with α-receptor blockade of sympathetic activity, suggesting that redistribution of blood flow aided by the sympathetic nervous system is a major determinant of critical oxygen extraction.63 Vasoconstriction after activation of the sympathetic nervous system during hypovolemia (hemorrhage) occurs in the arteries and medium-sized arterioles but not in terminal arterioles, which may even dilate, as judged from vital microscopy studies in animals.¶ Relatively spared terminal arteriolar blood flow is presumably caused by vasodilating metabolic responses to a decline in nutrient blood flow. Nevertheless, capillary flow usually diminishes, and heterogeneity, both in space and time, increases, particularly in irreversible shock and independent of cardiac output. Vasoconstriction is not confined to arteries, but also occurs in the venous vasculature, more in large than in small venules and particularly in the splanchnic area, and, again, this is largely mediated by increased activity of the sympathetic nervous system and vasopressin and angiotensin II release.5,12,68,108 Because most of the circulating blood volume is located in small venules, splanchnic venoconstriction results in a decrease in compliance and less volume for a given intravascular pressure in the venous system, increasing return of blood to the heart.5,7 Hence, partitioning circulating volume in stressed and unstressed portions now favors the former. During hypovolemic shock, the precapillary to postcapillary resistance increases, resulting in a decrease in capillary hydrostatic pressure and in fluid resorption from the interstitial space as opposed to normal filtration from capillary to interstitium, even though interstitial hydrostatic pressure decreases.4,5 This is accompanied by diminished transport of protein from blood to interstitium.124 Cellular water is mobilized, unless, at a later stage, the cell swells following Na+ overload.21,98,110,125–129 Studies on fluid volumes in hypovolemic shock are not equivocal, but generally suggest that the interstitial and cellular compartments are depleted in defense of the circulating blood volume to promote venous return to the heart.5,7,125,126 Mobilization of fluid from the interstitial and cellular compartment can be promoted by plasma hyperosmolarity, through an increase in the glucose concentration.130,131 Chronically starved rats with depleted glycogen stores more rapidly die of hypovolemic shock than fed ones, and this can be prevented by prior glucose infusion.130 In addition, the lymphatics may show increased pumping ability, increasing return of fluid into the systemic circulation independently of the reduced capillary fluid filtration rate.132 Lymphatic return of interstitial protein and fluid may contribute to repletion of circulating protein and fluid volume.132 Hemorrhage and hypovolemic shock lead to a decrease in hematocrit and a decrease in plasma proteins through transfer of fluid (and protein) from the interstitial to the intravascular space.4,5,124 Refilling of the intravascular space diminishes in time after a sudden decrease in circulating volume, when a decline in colloid osmotic pressure, associated with hypoproteinemia, and an increase in hydrostatic pressure accomplish a new steady state in capillary exchange through readjustment of the pericapillary hydrostatic and colloid osmotic pressures, which determine fluid and protein transport.5 Conversely, hypoproteinemia can promote transcapillary fluid transport and expansion of the interstitial space, if hydrostatic pressure returns toward normal (e.g., during crystalloid fluid resuscitation).126,133–136 During a sudden decrease in circulating blood volume by hemorrhage, some time is needed before the decrease in hematocrit and of proteins in blood is completed, and this decrease is aggravated by nonsanguineous fluid resuscitation.126,134,137 Finally, increased sympathetic discharge results in contraction of the spleen, releasing red blood cells into the circulation and defending a fall in hematocrit.14 During hypovolemic shock, the oxygen lack in the tissues causes a decline in the mitochondrial production and concentration of high-energy phosphates in the tissues because of greater breakdown than production of these compounds.24,29,46,138,139 This decline is a function of the severity and duration of regional hypoperfusion relative to oxygen demand. The decrease in the redox status and high-energy phosphates during experimental hypovolemic shock is more pronounced in some tissues (diaphragm, liver, kidney, and gut) than in others (heart and skeletal muscle), so regional lactate production may vary.* A decrease in high-energy phosphates heralds irreversible cell injury during ischemia, whereas a less severe decline may result only in prolonged programmed cell death—apoptosis. In animals with hypovolemic shock and in critically ill patients, the circulating levels of ATP can be diminished, and ATP degradation products, including adenosine, inosine, hypoxanthine, and xanthine, can be elevated, suggesting breakdown of ATP following a lack of oxygen in the tissues.28,29,56,141,142 Conversely, reperfusion is associated with restoration of energy charge, depending on the effect of ischemia, the oxygen demand, and the level of reperfusion. The intravenous administration of energy in the form of ATP-MgCl2 may help tissues (kidney, liver, heart, gut) to recover from ischemia and resume function, independently of the vasodilating effects of the compound.24,128,143,144 Also, pretreatment with coenzyme Q10, involved in the respiratory chain reactions in mitochondria, has a beneficial effect during hypovolemic shock and resuscitation, at least in dogs.81 Nevertheless, part of the mitochondrial dysfunction after trauma and hypovolemic shock has been suggested to be independent of a lack of oxygen.53 Near-infrared spectroscopy, which can be applied in animals and patients, may indeed reveal normal absorption spectra for tissue oxyhemoglobin and low mitochondrial cytochrome aa3 redox status.53,123,145 About 60% of the energy produced by respirating mitochondria is needed to fuel the Na+/K+ pump of the cell, through which the gradient in electrolyte concentrations and electrical potential over the cell membrane are controlled.24 When ATP becomes insufficient because of a decline in production associated with lack of oxygen and production of protons increases, the Na+/K+ pump is inhibited and the Na+/H+ exchanger is activated, and this results, together with a possibly selective increase in cell membrane permeability for ions, in an influx of Na+ into and efflux of H+ and K+ out of the cell, leading to cellular uptake of fluid.† Measurement of membrane potentials of skeletal muscle and liver in experimental animals has shown that hypovolemic shock rapidly decreases the transmembrane potential (a less negative inner membrane potential), associated with electrolyte and fluid shifts across the cell membrane.‡ A decrease in activity of the Na+/K+ pump may contribute to hyperkalemia because of potassium exchange between cells, interstitial fluid, and vascular space.38,46,111,125 Finally, calcium (Ca2+) influx into cells and their mitochondria inhibits cellular respiration and ultimately contributes to cellular damage and swelling, particularly during resuscitation, and this can be prevented by administration of Ca2+ antagonists.* Because of cellular influx, the plasma-free Ca2+ levels may decrease in experimental and human hypovolemic shock.127,149,150 Intracellular lysosomes lose their integrity so that proteolytic enzymes are released and contribute to cell death.4,24,107,151 These enzymes eventually may reach the systemic circulation and may damage remote organs.4,24,107,151 As has become apparent in past years, the cellular response to stress, such as heat and tissue hypoxia, involves the expression of certain genes, coding for synthesis of the so-called heat-shock proteins, which play an important role in protecting the cells against stress.152–155 The clinical significance of these molecular cellular changes is unknown. The response may be partially responsible, however, for the decreased susceptibility to and tissue injury by hemorrhagic shock in animals with a prior challenge by endotoxin or other forms of preconditioning.113,156 According to Starling’s law of the heart, a change in preload, approximated by the end-diastolic volume and determined by the venous return of blood to the heart, directly results in a change in stroke volume, defining myocardial function.7 The relationship between end-diastolic filling pressure and volume reflects compliance. Apart from preload, cardiac output also depends on afterload, which is approximated by the end-systolic volume of the heart, and contractility, reflected by the peak systolic pressure-to-volume relationship (maximal elastance).7,55,157 A diminished response of the stroke work by the heart, that is, the product of stroke volume and arterial blood pressure, to an increase in preload during resuscitation from hypovolemic shock may indicate diminished cardiac contractility that is associated with a worse outcome (e.g., caused by preexisting cardiac disease, hypovolemic shock itself, myocardial contusion, or combinations).4,55,158,159 The effect of hypovolemic shock on myocardial function in animal models is controversial. Depending on models, methods, and definitions of cardiac dysfunction, some authors describe a decrease, but others describe an unchanged function of the left side of the heart.† The latter can be explained if a decrease in contractility of the heart is masked by the inotropic effect of catecholamines and other positive inotropic substances, such as endothelin, liberated during hypovolemic shock, even though receptor-mediated catecholamine responses may decline.4,23,100,160 Although coronary blood flow may be defended, and the oxygen demands of the heart may decrease associated with a decrease in filling (preload) and arterial blood pressure (afterload) during initial hypovolemic shock, hypotension may become so severe that coronary vasodilation to compensate for a decline in perfusion pressure becomes exhausted, so that myocardial oxygen delivery decreases to less than the oxygen needs of the heart and ischemia ensues, particularly if tachycardia is present.3,21,140,161–163 This sequence leading to ischemia may occur primarily in endocardium because of more rapidly exhausted vasodilation in endocardium than epicardium and redistribution of blood flow from the inner to the outer layer of the heart.140 The subendocardium may become ischemic, and patchy necrosis may ensue. Because of regional transmural and intramural differences in vasodilator reserve, myocardial ischemia may be heterogeneously distributed and associated with a diminished redox state, lactate production, and creatine phosphate breakdown.140,164 Ischemia ultimately may contribute to a decrease in myocardial contractility during hypovolemic shock. Smooth muscle–dependent and, particularly, endothelium-dependent coronary vasomotion may be impaired after hypovolemic shock.89,165 Myocardial edema and compression of capillaries with resultant impairment of diffusion and extraction of oxygen may also contribute to a decrease in regional coronary blood flow, regional myocardial ischemia, and decreased myocardial function in hemorrhaged animals.127,131,140,161 Hypovolemic shock may induce a decrease in left ventricular compliance and relaxation.160,161 The diastolic dysfunction may be particularly pronounced during resuscitation from hypovolemic shock.157,160,161 Postischemic failure (stunning) also may play a role during resuscitation, at least temporarily. Ischemia-reperfusion of the heart results in accumulation of intracellular Ca2+.127 This may impair mitochondrial and sarcoplasmic reticulum function and contribute to impaired cardiac function after hypovolemic shock.127,160 In dogs, the administration of Ca2+ blockers may prevent such deterioration during resuscitation from hypovolemic shock.127 Finally, systemic release or intramyocardial production of negative inotropic substances and inflammatory mediators such as tumor necrosis factor (TNF)-α, interleukin (IL) 6 and platelet activating factor, oxidant damage, metabolic acidosis, diminished adrenoreceptor density, and resultant diminished sensitivity of the heart to circulating catecholamines may contribute to myocardial dysfunction during hypovolemic shock.* Pentoxifylline may improve endothelial and myocardial function.166 Reversibility of dysfunction is associated with survival.161 The clinical evidence for myocardial dysfunction during hypovolemic shock is scarce.46,55,163 Nevertheless, it is conceivable that severe hypotension reduces the balance between oxygen delivery and demand of the heart because many patients with hypovolemic shock may be elderly with coronary artery disease, compromising coronary vasodilation. Some may have preexisting impaired function while on beta blockers. For a patient with hypovolemic shock, a decrease in left ventricular compliance, contractility, or both may imply that a relatively high filling pressure would be needed to restore cardiac output during fluid resuscitation.55,71,161,163,171 The averaged optimal pulmonary capillary wedge pressure (PCWP), that is, the pressure above which cardiac output does not increase further, may not be elevated in patients with hypovolemic shock (i.e., 12 to 15 mm Hg), although in some patients, abnormally elevated filling pressures may be needed to increase cardiac output, or cardiac output does not increase at all during fluid resuscitation.55,71,171,172 A diminished function of the heart may hamper restoration of oxygen delivery to the tissues during resuscitation necessary for survival.9,45,55,159,160 Myocardial dysfunction may thus be greater in nonsurvivors than in survivors. There may be some electrocardiographic or enzymatic evidence for myocardial ischemia and injury, and some patients may experience a myocardial infarction as a complication of severe hypovolemic shock after hemorrhage.163,173 Hypovolemic shock often induces an increase in ventilatory minute volume, resulting in tachypnea or hyperventilation and a decrease in arterial PCO2.33,49,50,52,174 Unless complicated by pulmonary abnormalities, these changes are, at least initially, not the result of hypoxemia but an increase in dead-space ventilation following a decrease in pulmonary perfusion so that a higher minute ventilation is necessary for a given CO2 production to eliminate CO2 from the blood and to maintain a normal PCO2 in arterial blood.33,49,50 Minute ventilatory volume may increase further if a decrease in PCO2 is necessary to compensate for metabolic acidosis after accumulation of lactate in the blood.* The imbalance between increased demands of the diaphragm and reduced blood flow in shock may finally lead to respiratory muscle fatigue and a subsequent decline in ventilatory minute volume.50 Hypovolemic shock caused by trauma and hemorrhage and followed by extensive transfusion therapy of red blood cell concentrates can be complicated by pulmonary edema and impaired gas exchange.51,175–180 In some patients, fluid overloading, overtransfusion, and an elevated filtration pressure (PCWP) may be responsible: transfusion-associated circulatory overload (TACO). In others, pulmonary edema may be due to a pulmonary vascular injury, however, and increased vascular permeability at a relatively low PCWP, indicating noncardiogenic permeability edema or ARDS.51,174,177,178 The reaction to diuretics may help to differentiate between hydrostatic and permeability edema of the lungs. The latter seems relatively rare in polytransfused, polytraumatized patients unless associated with complications, but other studies suggest that about 30% of patients with severe trauma/hemorrhage, particularly if polytransfused, may develop ARDS.177,178,180,181 Experimental studies are at variance concerning alterations in capillary permeability of the lungs during hypovolemic shock and resuscitation.4,133,174,182,183 According to some investigators, hypovolemic shock following bleeding and transfusion mildly increases transvascular filtration of fluid and proteins and results in accumulation of interstitial fluid as a consequence of increased permeability,182 but other authors do not observe such changes.131,133,182,184 In other animal studies, however, traumatic/hypovolemic shock resulted in extensive morphologic changes of the lung, with endothelial and interstitial edema, accumulation of degranulated neutrophils, and scattered fat emboli, which may resemble the pulmonary changes after traumatic/hypovolemic shock in humans.156,174,185–187 As measured by the transvascular albumin flux in the lungs, almost 80% of patients with multiple trauma may show increased pulmonary vascular permeability in the disease course.51 This leak ultimately may contribute to pulmonary edema, impaired mechanics, and gas exchange.51 As suggested by animal experiments, among others, several factors may play a role, including release of proinflammatory mediators (TNF-α) and priming and activation of blood neutrophils after ischemia-reperfusion, contusion or ischemia-reperfusion of the lungs themselves, pulmonary microemboli of neutrophils, platelets and fat particles from the medulla of fractured long bones and pelvis, and neutrophilic antibodies or humoral or cellular breakdown products and released cytokines in long-stored and transfused blood products (transfusion-related acute lung injury, TRALI).* Translocated endotoxin may also play a role.185 Finally, aspiration of foreign material or gastric contents and posttraumatic pneumonia and sepsis may contribute to the development of ARDS in trauma patients. When pulmonary edema has developed, active resorption by alveolar cells becomes necessary for clearance. This process is cylic AMP–dependent and can be disturbed by inducible nitric oxide synthase (iNOS)–derived NO and peroxynitrite and enhanced by expression of heme oxygenase, which may mitigate lung injury in animal models.156 How this translates clinically is unclear. Classically, brain perfusion and microcirculation are considered to be relatively spared during progressive hypovolemia because of the extensive autoregulatory capacity of cerebral arteries.15,189 In case of autoregulation impairment after neurotrauma, however, brain perfusion may decrease, and subsequent reperfusion may contribute to secondary cerebral damage during hypovolemic shock and resuscitation. Hemorrhagic shock and resuscitation per se may also impair autoregulatory capacity of brain vessels, however, because of endothelial dysfunction and diminished NO-dependent vasodilator reactivity, so that the brain may experience an oxygen debt and subsequent metabolic and functional deterioration.29,88 Hypovolemic hypotension is an important risk factor for acute kidney injury and failure after trauma.138 During a decrease in cardiac output following progressive hemorrhage, renal blood flow can be maintained because of renal vasodilation, so that the kidneys may not participate in the systemic vasoconstriction that characterizes hypovolemic shock.3 Vasodilating prostaglandins are released in the kidney through activation of the cyclooxygenase pathway of arachidonic acid metabolism in response to ischemia, increased sympathetic activity, and angiotensin II, so that renal vasodilation during the early phase of hemorrhage can be blocked by prostaglandin synthesis inhibition, resulting in a profound decrease in blood flow even if accompanied by an increase in arterial blood pressure.3 When blood pressure decreases during progressive hypovolemia, the renal vessels constrict, impairing blood flow to the kidneys more than to other organs.* This is partly caused by a baroreflex-mediated increase in sympathetic activity; activation of the renin-angiotensin-aldosterone system; and release of catecholamines, angiotensin II, endothelin, and vasopressin.13,14,74 During prolonged hypovolemic shock, sympathetic inhibition may protect against renal ischemia.14 This propensity for vasoconstriction is thus partly offset if NO and other factors with vasodilatory actions are released intrarenally.4,86 Inhibition of NO synthesis increases blood pressure, however, and increases renal perfusion and glomerular filtration during hypovolemic shock.86 In another study, endothelium-dependent renal vasodilation was impaired after hypovolemic shock.87 Renal ischemia results in a decrease in glomerular filtration (prerenal renal failure) that is less than the decline in blood flow so that the filtration fraction often increases.138 The latter is caused by greater constriction of efferent than of afferent arterioles in glomeruli, in which high levels of circulating angiotensin II are probably involved. The decrease in glomerular filtration together with an increase in tubular resorption of electrolytes and fluids, mediated by increased levels of antidiuretic hormone released by the pituitary and decreased levels of atrial natriuretic peptides through low atrial filling, results in oliguria or anuria (<0.3 mL/kg/hour) and a low sodium content of urine.138 The decrease in renal perfusion during hypovolemic shock is often accompanied by redistribution of blood flow from outer to inner cortex and medulla, which is already borderline hypoxic even in the normal state.127 If long-lasting and severe, the cortical kidney becomes ischemic, despite a decrease in oxygen needs associated with fewer energy needs for tubular resorption in the presence of less filtration, so that the levels of high-energy phosphates decline.138,139 Severe and prolonged renal ischemia and metabolic deterioration finally result in acute kidney injury and failure with morphologic changes, particularly in proximal tubules and medullary segments (acute tubular necrosis) when an increase in renal perfusion does not immediately restore filtration and diuresis, but rather injures renal structures (reperfusion injury), limiting a return of blood flow and glomerular filtration during resuscitation.129,138,185,186 This is often recognized by a persistent oliguria and a gradual increase in creatinine and urea levels in blood. In addition, the plasma levels and urinary excretion of biomarkers of injury and dysfunction may increase.104 During hypovolemic shock, blood flow from stomach to colon is redistributed to other organs, and this may be primarily mediated by elevated sympathetic activity and increased levels of vasopressin and angiotensin II even though vascular reactivity to the latter may diminish.† Vasoconstriction may overwhelm NO and other vasodilating mechanisms, and endothelium-dependent vasodilation may be impaired after oxidant endothelial injury.194 Gut ischemia is aggravated further by the countercurrent mechanism in mucosal (villous) blood flow, promoting diffusional shunting of oxygen from arteries to veins, bypassing tissues. Other studies reported that gut mucosal blood flow may be relatively spared during hypovolemia, however.75,105 Portal blood flow decreases, and portal blood levels of lactate increase after gut ischemia.27,48,195 Gastric mucosal ischemia may result in diminished energy-consuming acid production and may predispose to mucosal stress ulceration.192,196 Microscopic studies in experimental animals show damage of gastric mucosa, villous epithelium in small bowel, and mucosa of the large bowel after hypovolemic shock.114,192,197–199 Gastric mucosal ischemia-reperfusion injury after bleeding may be aggravated by gastric acid itself, neutrophils, inflammatory mediators, endothelin, reactive oxygen species (ROS), and proteases.198,200 Bowel ischemia and mucosal damage during hypovolemic shock in the dog may ultimately lead to leakage of fluid from the bloodstream to the bowel lumen, instead of normal resorption of luminal fluids.115,197 Diarrhea may contribute to intravascular volume depletion during severe and prolonged hypovolemic shock, at least in animals. Gut mucosal ischemia, energy depletion, injury, and inflammation may compromise the barrier function of the mucosa, enhancing the likelihood that bacteria and endotoxins in intestinal lumen (large bowel) translocate through the damaged gut wall to lymph nodes, portal venous blood, or both.* The gut epithelial (lumen to plasma) permeability for small molecules also is increased. Indigenous flora, generated toxic ROS, cytokines, Ca2+ overload, iNOS, peroxynitrite, phospholipase A2 activation, and activated and adhering neutrophils during ischemia and reperfusion probably all play a role in the injury, promoting hyperpermeability and translocation.199,203 Mucosal injury and translocation can be inhibited by compounds targeted against these factors.199 Impaired detoxifying capacity of the Kupffer cells of the liver because of ischemia or preexistent liver disease may contribute further to bacteria and endotoxins reaching the systemic circulation and contributing to progression of shock by triggering an inflammation cascade, ultimately resulting in release of vasoactive substances.4,204,205 This translocation has been shown to contribute to the lethality of hypovolemic shock in experimental animals because clearance or blockade of translocated bacteria and endotoxins is associated with survival, and germ-free animals survive an episode of bleeding more often and longer than ones with normal intestinal flora.4,143,203,205 Finally, it has been shown that the absorptive capacity of the gut for carbohydrates, amino acids, and lipids decreases during hypovolemic shock.148,195 Although enteral feeding during hypovolemic shock and after resuscitation may increase metabolic demands of the gut, there is experimental evidence that luminal application of nutrients, particularly of enterocyte-fueling glutamine, induces an increase in small vessel blood flow, ameliorates damage, and diminishes the likelihood for translocation of endotoxins and bacteria during resuscitation from hemorrhage.206
Hypovolemic Shock
Pathogenesis and Pathophysiology
Circulatory Changes
General Changes
Oxygen Balance
 Treatment of hypovolemic shock, by infusing fluids and blood, is aimed at an increase in cardiac output and the oxygen content of blood and in oxygen delivery above the critical value so that oxygen uptake increases to meet body requirements and the lacticacidemia decreases.*
Treatment of hypovolemic shock, by infusing fluids and blood, is aimed at an increase in cardiac output and the oxygen content of blood and in oxygen delivery above the critical value so that oxygen uptake increases to meet body requirements and the lacticacidemia decreases.*
Macrocirculation
Microcirculation
 Traumatic/hypovolemic shock may induce expression of adhesion molecules on primed neutrophils and vascular endothelium and this, together with a reduced flow rate, may promote adherence of neutrophils to endothelium.95,111–119 This adherence may impair red blood cell flow, particularly in capillaries and postcapillary venules.
Traumatic/hypovolemic shock may induce expression of adhesion molecules on primed neutrophils and vascular endothelium and this, together with a reduced flow rate, may promote adherence of neutrophils to endothelium.95,111–119 This adherence may impair red blood cell flow, particularly in capillaries and postcapillary venules. Other authors suggest that capillary leukostasis is pressure-dependent and not receptor-dependent and reversible when perfusion pressure has been restored.121 Finally, endothelial cells may swell and may hamper capillary red and white blood cell flow.95,98,110,122 The microcirculation can be visualized, even in humans, by buccal or sublingual orthogonal polarization spectroscopy and side stream dark-field imaging.123
Other authors suggest that capillary leukostasis is pressure-dependent and not receptor-dependent and reversible when perfusion pressure has been restored.121 Finally, endothelial cells may swell and may hamper capillary red and white blood cell flow.95,98,110,122 The microcirculation can be visualized, even in humans, by buccal or sublingual orthogonal polarization spectroscopy and side stream dark-field imaging.123
Cells
Organ Perfusion and Function in Shock
Heart
Lung
Brain
Kidney
Gut
Related posts:

Full access? Get Clinical Tree


Hypovolemic Shock