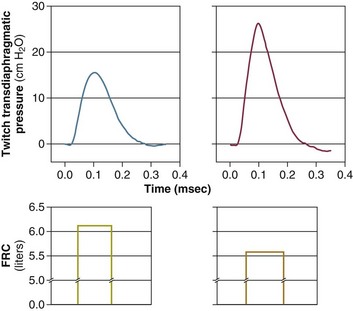
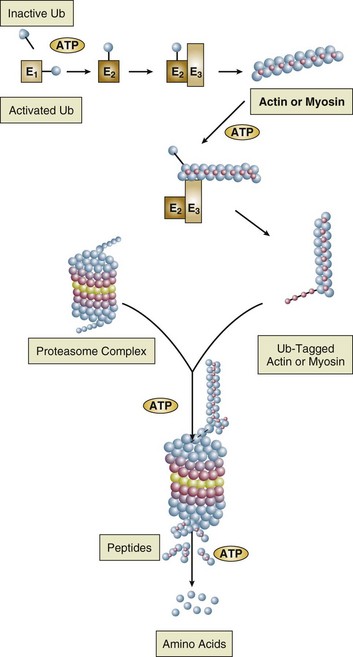
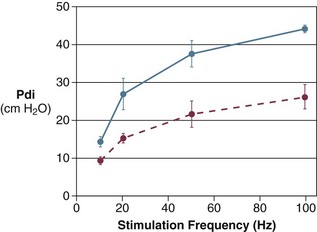
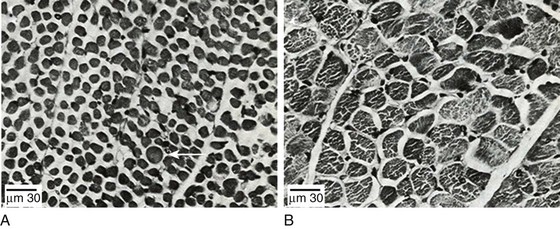
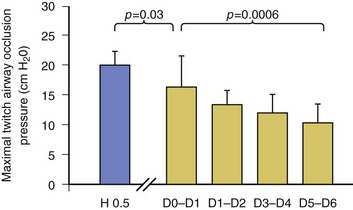
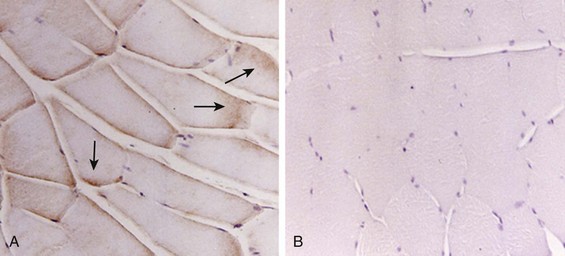
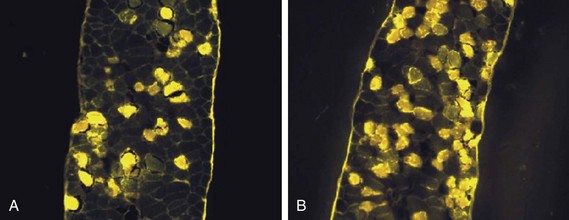
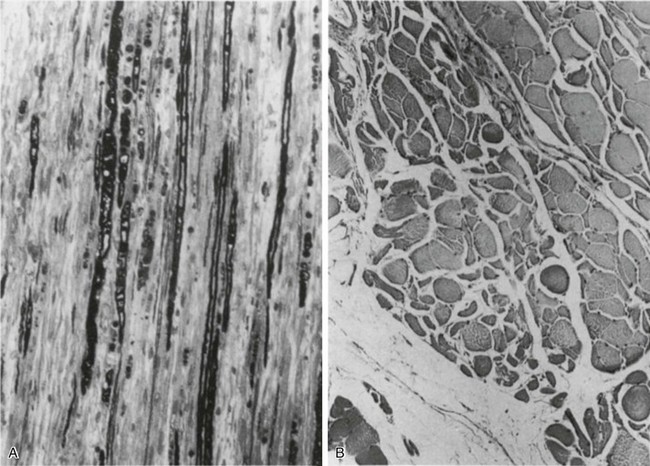
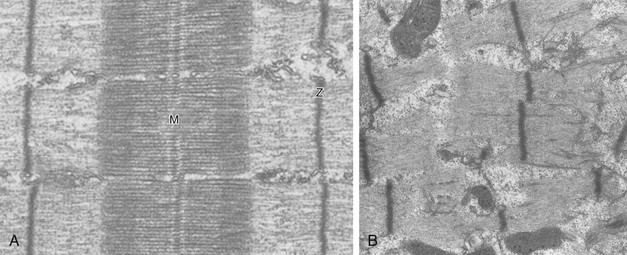
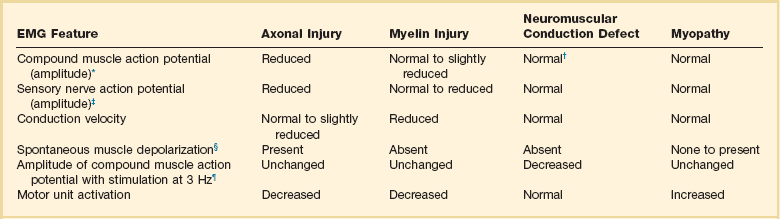
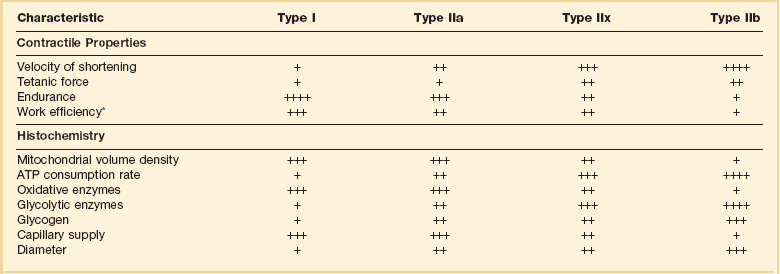
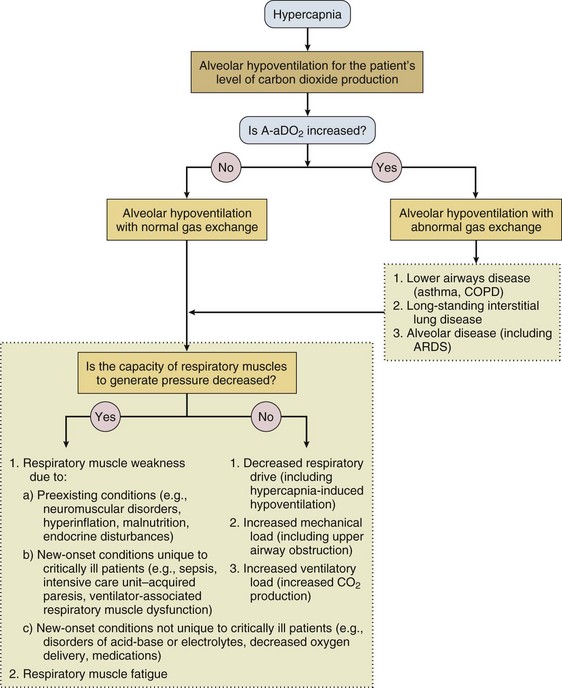
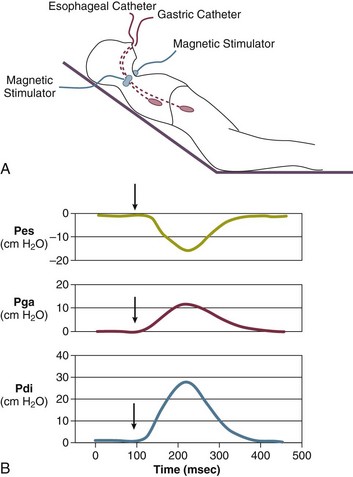
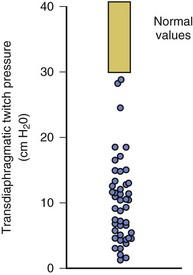
40 Hypercapnic respiratory failure is conventionally defined as a state in which ventilation is insufficient to maintain an arterial tension of carbon dioxide (PaCO2) of less than 45 mm Hg for the level of metabolic activity (measured by CO2 production, in which K (usually stated as 0.863) is a constant that converts measurement of where Hypercapnia is synonymous with alveolar hypoventilation for the patient’s level of carbon dioxide production. Alveolar hypoventilation can result from decreased neuromuscular capacity and increased respiratory load (Fig. 40.1).2 Isolated impairments in gas exchange—i.e., conditions in which the alveolar-arterial O2 gradient* is increased—usually do not cause hypercapnia.3 When gas exchange is impaired, however, the magnitude of neuromuscular derangements causing hypercapnia or the magnitude of increased respiratory loads causing hypercapnia is less than the corresponding derangements when gas exchange is normal. Conditions in which gas exchange contributes to hypercapnia are usually characterized by ventilation-perfusion inequality. When gas exchange is impaired total minute ventilation may actually be increased despite a concurrent decrease in alveolar ventilation. Isolated decreases in respiratory drive can produce ventilatory failure without distress. Potential causes include sedative overdose,1 hypothyroidism,4 metabolic alkalosis,5 semistarvation,6 and central alveolar hypoventilation syndrome. The latter can be either idiopathic (i.e., primary alveolar hypoventilation, or Ondine’s curse) or secondary to neurologic lesions, such as trauma, infection (poliomyelitis), infarction, and obesity hypoventilation syndrome.4 In mechanically ventilated patients, decreases in respiratory drive are suspected when patients develop marked increases in PCO2 despite relatively normal measurements of resistance and elastance and no apparent evidence of lower motor neuron disease.7 Whether sleep deprivation decreases respiratory drive remains controversial.8,9 Measurements of airway pressure during maximal voluntary inspiratory efforts are used to evaluate global inspiratory muscle strength.10 In healthy subjects, maximum inspiratory airway pressure is usually more negative than −80 cm H2O.10 In mechanically ventilated patients recovering from an episode or acute respiratory failure, maximum inspiratory airway pressure can range from less negative than −20 cm H2O to about −100 cm H2O.7,11,12 Values of maximal airway pressure during voluntary maneuvers depend greatly on a level of motivation and comprehension of the maneuver (often not obtainable in critically ill patients).13 Not surprisingly, in patients requiring short-term mechanical ventilation, measurements of maximum inspiratory airway pressure commonly do not differentiate between weaning success and weaning failure patients.12–14 In contrast to the voluntary nature of maximal voluntary inspiratory efforts, transdiaphragmatic pressures elicited by single stimulations of the phrenic nerves—or twitch pressure—are independent of patients’ motivation and eliminate the influence of the central nervous system.10 Activation can be achieved with either an electrical stimulator15 or a magnetic stimulator,15 though the latter is easier to use in mechanically ventilated patients (Fig. 40.2).13,16,17 In healthy volunteers, magnetic stimulation elicits twitch pressures that average 31 to 39 cm H2O.10 In patients with severe chronic obstructive pulmonary disease (COPD), twitch pressures average 19 to 20 cm H2O.18,19 The value of transdiaphragmatic twitch pressure in patients recovering from an episode of acute respiratory failure is about one third of that recorded in healthy subjects (Fig. 40.3).20 This marked reduction in twitch pressure16,17 indicates the presence of respiratory muscle weakness in most of these patients. Respiratory muscle weakness in critically ill patients can result from preexisting conditions or from new-onset conditions. Neuromuscular diseases, malnutrition, endocrine disorders, and hyperinflation are some of the preexisting conditions that can cause respiratory muscle weakness. The existence of preexisting conditions can be recognized before the patient develops acute ventilatory failure, at the time the patient develops acute ventilatory failure, or when acute ventilatory failure is already established.4,21–24 The capacity of the respiratory muscles to generate tension can be decreased in certain disorders, such as stroke, amyotrophic lateral sclerosis, spinal cord injuries, poliomyelitis, Guillain-Barré syndrome, neuropathies due to massive intoxications of arsenic25 or thallium,26 chronic inflammatory demyelinating polyneuropathy, axonopathy of acute intermittent porphyria, myasthenia gravis, acid maltase deficiency, and muscular dystrophies such as myotonic dystrophy (i.e., the most frequent adult form of muscular dystrophy).4,27,28 Hypercapnic respiratory failure usually occurs when respiratory muscle strength falls to 39% of the predicted normal value.29 However, Gibson and associates30 described several patients with neuromuscular disease who had a normal partial pressure of CO2 despite decreases in respiratory muscle strength to less than 20% of predicted. Conversely, some patients with only moderate respiratory muscle weakness displayed hypercapnia (Fig. 40.4).30 In other words, reductions in muscle strength do not consistently predict alveolar hypoventilation in this setting. Hyperinflation is a common preexisting problem in patients with obstructive lung diseases such as COPD,4 cystic fibrosis,31 bronchiolitis,32 and lymphangioleiomyomatosis.4 The severity of preexisting hyperinflation commonly worsens in patients experiencing an exacerbation of COPD.33 Hyperinflation has a number of adverse effects on inspiratory muscle function: the inspiratory muscles operate at an unfavorable position of the length-tension relationship (Fig. 40.5);34 flattening of the diaphragm decreases the size of the zone of apposition with the result that diaphragmatic contraction causes less effective rib cage expansion.4 Hyperinflation also has an adverse effect on the elastic recoil of the thoracic cage.4 This means that the inspiratory muscles must work not only against the elastic recoil of the lungs but also against that of the thoracic cage. The functional consequences of dynamic hyperinflation are probably the main mechanisms of ventilatory failure in patients with COPD.35 Hyperinflation can also occur de novo in patients with pneumonia, chest trauma, and acute respiratory distress syndrome (ARDS).33,36 Impairment of inspiratory muscle function is less likely in patients with ARDS than in patients with obstructive lung disease because patients with ARDS breathe at a low lung volume despite dynamic hyperinflation.36,37 Malnutrition is highly prevalent among critically ill patients requiring mechanical ventilation,38,39 and it is associated with poor prognosis.39 Malnutrition decreases muscle mass and respiratory muscle strength both in humans40,41 and in laboratory animals.42–44 Likely mechanisms contributing to decrease in respiratory muscle mass include proteolysis of myofibrillar proteins by the ubiquitin-proteasome proteolytic system (Fig. 40.6) and apoptosis.4,45 Apoptosis can be triggered by local and circulating tumor necrosis factor-α and by release of cytochrome c from the mitochondria.46,47 In patients with COPD, inspiratory muscle strength is about 30% less in poorly nourished patients than in well-nourished patients with equivalent airway obstruction.48 Similarly, malnourished patients with anorexia nervosa can present with reduced inspiratory muscle strength to 35% to 50% predicted,41 impaired respiratory muscle endurance,49 impaired hypercapnic ventilatory response,49 and occasionally, with hypercapnia at rest.41 In malnourished patients, inspiratory weakness,41,48,49 fatigability,48 and dyspnea48 are partially reversible with nutritional support. The process is slow and, in laboratory animals, can take months of refeeding for muscle mass to return to normal values.50 To date, it remains unclear whether malnutrition by itself can cause sufficient respiratory muscle weakness to produce hypoventilation. It is more likely for malnutrition to be a contributory factor and not a sole cause of hypercapnic respiratory failure. Endocrine disturbances such as hypothyroidism,51 hyperthyroidism,47,52–54 and acromegaly55 can adversely affect respiratory muscle function. Proteolysis of myofibrillar proteins by the ubiquitin-proteasome proteolytic system45 (see Fig. 40.6) is probably responsible for respiratory muscle catabolism and weakness of hyperthyroidism.47 This mechanism is implicated in muscle wasting associated with acidosis, renal failure, denervation, cancer, diabetes, acquired immunodeficiency syndrome (AIDS), trauma, and burns.45 In contrast to other endocrine disturbances, respiratory muscle weakness is unusual in patients with Cushing syndrome.56 Since the late 1980s, several groups have studied the effect of mechanical ventilation on the muscles of laboratory animals.57 A seminal study by Anzueto and colleagues58 showed that 11 days of controlled mechanical ventilation (CMV) together with neuromuscular blocking agents produced a 46% decrease in respiratory muscle strength (Fig. 40.7). Subsequent studies have revealed that complete cessation of diaphragmatic activity with CMV—alone59 or in combination with neuromuscular blocking agents60—results in atrophy and injury of diaphragmatic fibers (Fig. 40.8). Muscle fibers generate less force in response to stimulation, not simply because of their decreased bulk but even when normalized for cross-sectional area.57 The decrease in diaphragmatic force ranges from 20% to more than 50%. The alterations in muscle function occur rapidly, within 12 hours of instituting mechanical ventilation,61 and they appear to increase as ventilator duration is prolonged.62,63 Mechanical ventilation may also harm the respiratory muscles of patients. In 1988, Knisely and coworkers64 reported that the cross-sectional area of diaphragmatic fibers in infants who had died after receiving mechanical ventilation for 12 or more days was much smaller than that in infants who had received mechanical ventilation for 7 days or less (Fig. 40.9). Fibers taken from extradiaphragmatic muscles were similar in the two groups. Twenty years later, Levine and associates65 obtained biopsies of the diaphragm from 14 brain-dead organ donors who were maintained on CMV for 18 to 69 hours. They also obtained intraoperative biopsies of the diaphragms of eight control patients who had received CMV for 2 to 3 hours. Histologic measurements revealed marked diaphragmatic atrophy in the brain-dead patients. Compared with the control group, the mean cross-sectional areas of muscle fibers were decreased by more than 50%. A number of these results have been corroborated by Jaber and colleagues63 and Hermans and coworkers,66 who also reported a progressive decrease in diaphragmatic contractility of mechanically ventilated patients. The decrease in contractility correlated with the duration of ventilator support (Fig. 40.10).63,66 Biochemical and gene-expression studies in animals and humans65,67 suggest that oxidative stress is probably one of the most proximal mechanisms in the biochemical cascade that leads to ventilator-induced muscle injury.68 Oxidative stress decreases contractility by causing protein oxidation and by promoting protein catabolism68—including up-regulation of the autophagy-lysosome pathway.69 The synergism between ventilator-induced muscle injury and oxidative stress has caused investigators to explore whether early administration of antioxidants might minimize muscle injury.70–72 In a study of more than 200 critically ill patients—80% of whom required acute ventilator support—duration of mechanical ventilation was nearly 3 days shorter in those who completed a 10-day antioxidant supplementation protocol (vitamin E and vitamin C) than in those who completed a 10-day course of placebo.73 Similar results have been reported in critically ill surgical patients requiring mechanical ventilation.74 Whether the decrease in duration of mechanical ventilation was, at least in part, due to the potential positive effects of antioxidants on the respiratory muscles remains to be demonstrated. Of concern, however, was the report that administration of the antioxidant N-acetylcysteine (NAC) to critically ill patients with severe sepsis worsened sepsis-induced organ failure.75 In animal models, ventilator settings can affect the extent of ventilator-associated respiratory muscle dysfunction.76–79 In rabbits, assist-control mechanical ventilation causes a nonsignificant decrease in diaphragm muscle contractility, which contrasts with the 48% decrease recorded with CMV.76 Similar results have been obtained when comparing CMV to adaptive support ventilation in piglets78 or when comparing CMV to pressure support in rats79 or CMV to intermittent spontaneous breathing again in rats.77 These observations raise the important question of whether maintenance of partial diaphragmatic activity or intermittent loading of the diaphragm could prevent the harm done to diaphragmatic function by mechanical ventilation.80 Considering that decrease in protein synthesis probably contributes to ventilator-associated respiratory muscle dysfunction,81,82 it would seem biologically plausible that administration of anabolic factors—such as growth hormone—might be of benefit in ventilated patients. Unfortunately, when growth hormone has been administered to patients requiring prolonged mechanical ventilation, duration of mechanical ventilation was not decreased, nor was muscle strength increased.83 Of concern was the report that recombinant growth hormone can increase the mortality rate of critically ill patients.84 An even more fundamental point in regard to ventilator-associated respiratory muscle dysfunction has been raised recently by the preliminary data of Hooijman and associates.85 These investigators measured the contractile properties of single diaphragmatic fibers obtained from nine brain-dead organ donors who had received CMV for 30 to 84 hours (case subjects) and from nine patients undergoing surgery for localized lung cancer (control subjects). In this preliminary study no difference was noted between the muscle fibers of the two groups of patients with respect to maximum force per unit area, calcium sensitivity of contractile force, and rate constant of redevelopment of force (Ktr). How to reconcile the preliminary results of Hooijman and associates85 with previous data obtained on muscle strips from animal models and by phrenic nerve stimulation in patients remains to be determined. Sepsis, a common occurrence in critically ill patients, can produce ventilatory failure by causing respiratory muscle dysfunction and increased metabolic demands.86 Septic animals develop failure of neuromuscular transmission (due to increased sarcolemmal electric potential)87–89 and failure of excitation-contraction coupling.86,90 Mechanisms responsible for failure of excitation-contraction coupling include the cytotoxic effect of nitric oxide and its metabolites, free radicals, ubiquitin-proteasome proteolysis, and possibly, decrease in nicotinic acetylcholine receptors.91,92 Local dysregulation of the circulation and Krebs cycle may also contribute.86 Nitric oxide, a free radical that has a negative inotropic effect in the heart and skeletal muscle, is produced in large amounts during sepsis by a nitric oxide synthase inducible by lipopolysaccharide and several cytokines (Fig. 40.11).93 Increased expression of inducible nitric oxide synthase in the diaphragm during sepsis is associated with morphologic evidence of widespread damage to the myofiber membrane, or sarcolemma (Fig. 40.12).88 Diaphragmatic contractions enhance this sepsis-induced sarcolemmal injury,94 while early mechanical ventilation decreases sarcolemmal injury and the associated diaphragmatic dysfunction.95 The beneficial effects of resting the diaphragm with the use of mechanical ventilation are not coupled with a decrease in oxidative stress or with a decrease in the expression of inducible nitric oxide synthase in the muscle.95 These observations suggest the existence of a detrimental interaction of two independent stressors (oxidative and biomechanical stresses) on the sarcolemma during sepsis.95 To determine whether the inducible nitric oxide synthase pathway contributes to impaired skeletal muscle contractility in humans, Lanone and coworkers96 obtained samples of the rectus abdominis in 16 septic patients and 21 control subjects. The muscles of the patients had lower contractile force, and increases in inducible nitric oxide synthase expression (mRNA and protein) and activity. Immunohistochemical studies revealed the generation of peroxynitrite (a highly reactive oxidant formed by the reaction of nitric oxide with superoxide anion). Exposure of control muscles to the amount of peroxynitrite found in patients caused an irreversible decrease in force generation. These data suggest that sepsis decreases muscle force through the production of nitric oxide and its toxic byproducts. Production of nitric oxide in sepsis may be protective and not solely deleterious.94,97–99 In mice deficient in inducible97 or constitutive (neuronal) nitric oxide synthase,94 endotoxin caused a greater decline in diaphragmatic contractility than in nondeficient mice. This finding contrasts with the observation that nitric oxide synthase inhibitors prevent muscle dysfunction in septic rats.88,93,100 Although the results may be species-dependent,94 the data underscore that nitric oxide has both antioxidant and pro-oxidant actions.101 In addition to nitric oxide and its derivatives, several other oxygen-derived free radicals (superoxide anion, hydroxyl radicals, hydrogen peroxide) contribute to the decreased contractility of the diaphragm in sepsis.99,102–104 This increased expression of oxygen-derived free radicals in sepsis is accompanied with enhanced activity of the antioxidant enzyme superoxide dismutase102 and with increased expression of the heme oxygenase-1 pathway.104 The heme oxygenase-1 pathway is a powerful cellular system that protects against oxidative stress and contractile fatigue during sepsis.104 Administration of an inhibitor (zinc protoporphyrin IX) or an inducer (hemin) of heme oxygenase activity respectively enhances or reduces the oxidative stress and contractile failure of the diaphragm in a rat model of sepsis.104 In septic rats, decreased diaphragmatic contractility can also be improved by the administration of specific scavengers of superoxide ions, hydrogen peroxide, and hydroxyl radicals.102 While cared for in the ICU, critically ill patients can develop muscle weakness and, occasionally, paralysis. Some of these patients have evidence for axonal degeneration and denervation atrophy (Fig. 40.13).4 This constellation of findings is known as critical illness polyneuropathy (Table 40.1).105 Cytokines106 and low-molecular-weight neurotoxins107 released during episodes of sepsis or when patients develop multiple organ failure are thought to be responsible for this axonal degeneration. Critical illness polyneuropathy has been considered one of the manifestations of multiple organ failure syndrome. Sepsis and multiple organ failure, though, are not essential prerequisites for the development of critical illness polyneuropathy.108,109 Tight control of hyperglycemia may reduce the risk of polyneuropathy and the duration of mechanical ventilation.110 Table 40.1 Examples of Electromyography (EMG) Findings in Respiratory Muscle Weakness *Elicited by motor nerve stimulation. †Decreased in Lambert-Eaton syndrome. ‡Elicited by sensory nerve stimulation. §Spontaneous muscle depolarization (caused by denervation) is detected by presence of fibrillation potentials and positive sharp waves. ¶Repetitive nerve stimulation is performed to exclude neuromuscular transmission defects such as prolonged neuromuscular paralysis. From Laghi F, Tobin MJ: Disorders of the respiratory muscles. Am J Respir Crit Care Med 2003;168:10. In other patients, rather than axonopathy, there is evidence of isolated myopathy (i.e., critical illness myopathy).4 Patients developing isolated myopathy often have been treated with steroids and neuromuscular blocking agents (e.g., patients with status asthmaticus).4 Muscle biopsies demonstrate a general decrease in myofibrillar protein content and a selective loss of thick filaments (myosin) within type I and type II fibers (Table 40.2 and Fig. 40.14). Animal models of critical illness myopathy suggest that medical denervation with paralytic agents cause an up-regulation of glucocorticoid receptors in the muscle.111 If the animal subsequently receives high-dose corticosteroids, it will develop depletion of thick myosin filaments.111 Although a decrease in thick-filament proteins may be important for prolonged weakness,112 this decrease is probably not the cause of the acute paralysis,113 particularly in patients with compound motor action potentials of low amplitude.114 Impaired muscle membrane excitability is probably more important during the acute stage.113,115 Table 40.2 Characteristics of Types of Muscle Fibers A single myosin heavy chain isoform typically is expressed within an adult skeletal muscle fiber. Fibers classified as type I, IIa, IIx, or IIb express myosin heavy chain isoform I (or slow), IIa, IIx, or IIb, respectively. Type IIx fibers have been reported in peripheral muscles of humans and animals and in the diaphragm of animals. Type IIx fibers have not been reported in the human diaphragm. More than one myosin heavy chain isoform is expressed in a few fibers (about 14% of adult rat diaphragm coexpresses myosin heavy chain isoforms IIb and IIx, and less than 1% coexpresses myosin heavy chain isoforms I and IIa).226 Whereas the velocity of muscle contraction depends primarily on the myosin heavy chain isoform, the velocity of muscle relaxation is mainly determined by troponin C calcium binding and release and by calcium reuptake by the sarcoendoplasmic reticulum calcium-adenosine triphosphatase (SERCA). Several SERCA isoenzymes have been identified: SERCA 1 is expressed in type II fibers (fast calcium reuptake), and SERCA 2a is expressed in type I fibers (slow calcium reuptake).227 The density of pumping sites largely accounts for different rates of calcium uptake in fast- and slow-twitch muscle fibers.227 Despite this separation of tasks, velocity of contraction and velocity of relaxation tend to parallel each other: type II fibers contract and relax with a greater velocity than type I fibers. Slower velocity of relaxation allows fusion of repetitive twitches at lower frequencies of stimulation than with fast relaxations. Impairment of SERCA activity has been implicated in the development of fatigue and in disease states including heart failure and corticosteroid myopathy. *Amount of work performed per unit of adenosine triphosphate (ATP) consumed. From Laghi F, Tobin MJ: Disorders of the respiratory muscles. Am J Respir Crit Care Med 2003;168:10. In the last few years it has become increasingly apparent that critical illness neuropathy and myopathy often coexist.106,109,115–118 It has become common to refer to patients who become weak while in the ICU as a result of acquired neuropathy or myopathy (not associated with a known disorder) as simply having ICU-acquired paresis.115,116,118 In patients with ICU-acquired paresis duration of weaning from mechanical ventilation can be two to seven times longer than in patients without ICU-acquired paresis.119
Hypoventilation and Respiratory Muscle Dysfunction
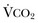 ).1 Under steady-state conditions, the relationship among PaCO2, alveolar ventilation (
).1 Under steady-state conditions, the relationship among PaCO2, alveolar ventilation (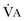 ), and
), and 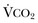 is given by the equation:
is given by the equation:
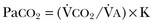
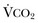 from standard conditions to body temperature conditions. The term
from standard conditions to body temperature conditions. The term 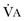 represents the portion of minute ventilation (
represents the portion of minute ventilation (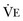 ) that reaches the terminal gas exchange units and is calculated as
) that reaches the terminal gas exchange units and is calculated as
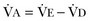
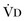 equals dead space ventilation. A reduction in
equals dead space ventilation. A reduction in 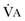 may result from an inadequate
may result from an inadequate 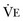 or an increase in
or an increase in 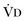 (resulting from an increase in true
(resulting from an increase in true 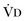 or a functional increase in
or a functional increase in 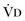 secondary to lung regions with high ventilation-perfusion [
secondary to lung regions with high ventilation-perfusion [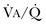 ] relationships).*
] relationships).*
Decreased Neuromuscular Capacity
Decreased Respiratory Center Output
Respiratory Muscle Weakness
Detection of Respiratory Muscle Weakness in Critically Ill Patients
Weakness Due to Preexisting Conditions
Neuromuscular Disorders
Hyperinflation
Malnutrition
Endocrine Disturbances
Weakness Due to New-Onset Conditions
Ventilator-Associated Respiratory Muscle Dysfunction
Sepsis-Associated Myopathy
Intensive Care Unit–Acquired Paresis
Related posts:

Full access? Get Clinical Tree


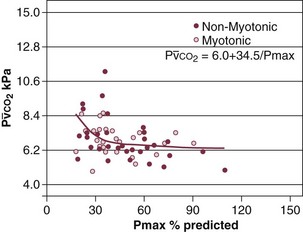
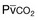 ) in patients with respiratory muscle weakness. Respiratory muscle strength is the arithmetic sum of maximum static inspiratory and expiratory mouth pressures (Pmax = P
) in patients with respiratory muscle weakness. Respiratory muscle strength is the arithmetic sum of maximum static inspiratory and expiratory mouth pressures (Pmax = P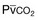 rose, although considerable variability was observed among patients. The regression lines were similar in the myotonic and nonmyotonic patients.
rose, although considerable variability was observed among patients. The regression lines were similar in the myotonic and nonmyotonic patients.