Soluble in aqueous medium
Stable in solution
Stable in air within the container
Stable when diluted
Stable in light
Stable at room temperature
Inexpensive
Potent enough to provide surgical anaesthesia without supplement
Induction in one arm–brain circulation time
High oil/water solubility
Analgesia
No pain on injection
Non-epileptogenic
No cardiac irritability
No cardiovascular depression
No respiratory depression
Muscle relaxation
No increase in intracranial pressure
Non-cumulative with infusion
Unaffected by renal and hepatic failure
Metabolites inactive and non-toxic
Non-teratogenic
Barbiturates
Examples – pentobarbital, phenobarbital, thiopental
Chemical structure
Barbiturates are based on barbituric acid, which contains the six-membered pyrimidine nucleus that is a building block of nucleosides. Oxypyrimidines may exist in two structural forms, enol and keto (Figure 30.4). Thiobarbiturates have a similar pair of structural forms. Chemically, barbituric acid is a condensation product (formation of the chemical bonds involves the removal of water molecules) of malonic acid and urea. Substitutions on atoms at positions 1, 2 and 5 confer and modulate hypnotic activity.
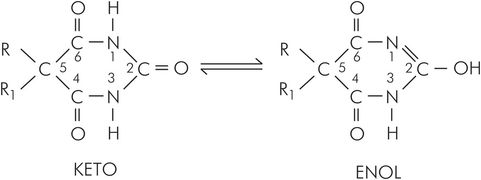
Basic chemical structure of barbiturates, showing enol and keto forms
Structure–activity relationship
Barbituric acid has no hypnotic activity. Clinically active barbiturates are created by the addition of various alkyl groups to C-5. Barbiturates act at the GABAA receptor to increase the effect and duration of action of GABA and also block AMPA/kainite receptors and inhibit glutamate release via the P/Q type high-voltage-activated calcium channels. Oxybarbiturates (e.g. phenobarbital) are the basic form of barbiturate, which have a slow onset of action and a long duration. They function well as basal hypnotics and sedatives but are too slow for use as anaesthetic agents. Replacing the oxygen on C-2 with sulphur produces a thiobarbiturate (e.g. thiopental), which has a rapid onset of action and a relatively short duration of action and recovery period. Replacing the hydrogen on N-1 of the basic oxybarbiturate with a methyl group produces a methylbarbiturate (e.g. methohexital). Methylbarbiturates also have a rapid onset and short duration of action plus a short recovery. However, they also produce some excitatory activity, which takes the form of sporadic uncoordinated movements during induction of anaesthesia. These may be mild or vigorous, but are usually distinguishable from the repetitive jerking of epileptiform convulsions, or the diffuse muscular shimmering (fasciculation) seen with suxamethonium. Modification of the alkyl groups on C-5 alters the potency of the drug. Oxy-, methyl-, and thio-barbiturates are all clinically useful, but the addition of both sulphur and a methyl group (methylthiobarbiturates) results in excessive excitatory activity, precluding the use of such a compound in the clinical setting.
The structural classification of barbiturates is shown in Figure 30.5.
Clinical effects
Phenobarbital and pentobarbital have a slow onset of action, and provide anxiolysis and anticonvulsant activity. The intravenously administered induction agent thiopental is the most relevant to anaesthetic practice: it is a water-soluble barbiturate agent that induces anaesthesia within one arm–brain circulation time of IV injection. It is highly lipophilic, and easily and rapidly crosses the blood–brain barrier to penetrate the brain.
Complications associated with barbiturate usage
Porphyria
The porphyrias are a group of diverse diseases of inborn or acquired errors of metabolism that result in the secretion of excessive amounts of porphyrins and precursors (Figure 30.6).
| Erythropoietic | Congenital erythropoietic porphyria |
| Hepatic | Acute intermittent porphyria Hereditary coproporphyria Variegate porphyria Porphyria cutanea tarda Toxic porphyria |
| Erythropoietic and hepatic | Protoporphyria |
Porphyrins comprise four pyrrole rings linked together to form a larger ring. They are fundamental constituents of haemoglobin, myoglobin, cytochromes and catalases, and all contain iron. Porphyria is very rare in the UK. While still rare, acute intermittent porphyria is more common in South Africa, Sweden and Finland, and variegate porphyria is more common in Sweden and Finland. These are both inherited by autosomal dominance, and are the most relevant to anaesthesia. They affect the liver, where haem is produced as a component of the cytochrome P450 enzyme, and therefore affect drug metabolism. Many drugs increase porphyrin production, and administration may result in neurotoxic porphyrin levels. Drugs to avoid include barbiturates, benzodiazepines and steroids, but propofol is thought to be safe.
Subcutaneous extravasation
Extravasation of thiopental causes localised pain and tissue damage due to the highly alkaline medium in which it is mixed. It does not usually cause major sequelae, but injection in proximity to a nerve may cause damage. The pain and bruising can be reduced by dilution with a small dose (10 mL) of saline, or 10 mL 1% procaine, which vasodilates as well as diluting the thiopental. The damage is due to the high pH, and is immediate, so any damage limitation measures should be performed as quickly as possible.
Intra-arterial injection
Intra-arterial injection of thiopental is a potentially serious complication, causing endothelial damage. This is related to the change in pH of the solution as it is diluted in the blood, which results in a precipitation of insoluble microcrystals that block the narrowing arterial tree – whereas in veins any such crystals are carried away and diluted so that they dissolve before causing any problems. The precipitate causes arterial intimal damage, local release of noradrenaline and the release of ATP from damaged red cells and platelets, which initiates vascular thrombosis. Clinically there is pain, with delayed or absent onset of sleep. Blanching or cyanosis of the affected area, loss of the peripheral pulse of the injected limb and gangrene may occur. Arterial thrombosis may gradually develop, progressing for up to 15 days. The treatment is as follows:
Management of suspected intra-arterial injection of thiopental
Stop the injection, but leave the needle or cannula in place.
Dilute immediately by injecting normal saline into the artery.
Administer local anaesthetic and/or vasodilator directly into the artery:
Lidocaine 50 mg (5 mL 1% solution)
Procaine hydrochloride 50–100 mg (10–20 mL 0.5% solution)
Phenoxybenzamine (α-adrenoceptor antagonist) 0.5 mg or infuse 50–200 μg min–1
Administer papaverine systemically 40–80 mg (10–20 mL 0.4% solution).
Consider sympathetic neural blockade (stellate ganglion or brachial plexus block). This produces prolonged vasodilatation to improve circulation and tissue oxygenation while improving clearance of the crystals.
Start IV heparin.
Consider intra-arterial injection of hydrocortisone.
Postpone non-urgent surgery.
Thiopental
Physical
Sodium thiopental is a thiobarbiturate. It is supplied in a rubber-topped bottle, as a pale yellow powder containing 6% anhydrous sodium carbonate (a base). The gaseous environment within the bottle is nitrogen. The sodium carbonate prevents CO2 in the air from forming free acid that would react with the thiopental. The sodium ion replaces the hydrogen ion that associates with C-1 and C-2 of the base compound, as shown in Figure 30.7.
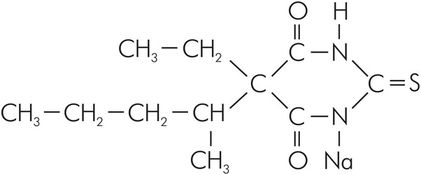
Structure of thiopental
The powder is readily soluble in water, producing a 2.5% (2.5 g 100 mL–1) solution with pH 11. At pH 11, having a pKa = 7.6, it is almost entirely (99.9%) ionised. However, once in the blood the pH falls towards 7.4, at which 61% of the drug is non-ionised. This non-ionised portion is the more lipid-soluble and readily crosses the blood–brain barrier into the lipid-rich brain tissue. Of the drug, 60–80% is reversibly bound to protein (mainly to albumin) and is therefore non-diffusible and inactive.
Clinical
IV injection of thiopental rarely causes pain, and venous thrombosis occurs in only 3–4% of patients. Characteristically, the subject may be aware of a taste of onions or garlic before the onset of sleep. The redistribution half-life of 4 minutes leads to rapid recovery.
Central nervous system
Thiopental causes cortical depression, with a plasma level of 40 μg mL–1 being typical for sleep. Rapid and smooth induction of anaesthesia occurs within one arm–brain circulation time with a low incidence (4%) of minor excitatory movements. Thiopental has no analgesic activity and may be antanalgesic. It is an anticonvulsant agent. During induction, the EEG shows the onset of high-amplitude waves of 10–30 Hz, followed by depression of cortical activity with an isoelectric picture accompanied by occasional bursts of 10 Hz activity.
Cerebral metabolic rate of oxygen consumption (CMRO2) is reduced, which leads to cerebral vasoconstriction with a concomitant fall in cerebral blood flow (CBF) and ICP. The fall in blood pressure that also occurs leads to a fall in CBF, with an opposite effect on cerebrovascular resistance. The reduction in ICP and CMRO2 are useful properties of thiopental for use in cerebral protection for situations such as cardiopulmonary bypass or standstill, and for the prevention of secondary brain damage in trauma.
A single large dose results in the return of wakefulness when the plasma level is higher than if smaller doses are used. This is termed acute tolerance. The technique of injecting a predetermined dose without slowly titrating it to effect allows the use of lower doses while still achieving satisfactory peak concentrations in the cerebral circulation and therefore the brain. A more rapid recovery results from a correspondingly more rapid fall in cerebral plasma thiopental concentration. Late recovery is also improved, as the total dose is lower.
Cardiovascular system
Thiopental causes a dose-dependent reduction in vascular tone with a reduction in SVR, CVP and PCWP. Preload and afterload are therefore both reduced, resulting in a decrease in mean arterial blood pressure and in left and right ventricular work. There is a slight compensatory increase in heart rate. High doses given rapidly can cause severe myocardial depression and hypotension, particularly in hypovolaemic patients or those receiving antihypertensive medication. Extreme caution should be exercised if thiopental is to be used in patients who cannot increase their cardiac output to compensate for a drop in vascular resistance, such as those with valvular stenosis or cardiac tamponade. Gradual administration can produce an isoelectric EEG without any significant effect on blood pressure or heart rate. Cardiac oxygen consumption is increased in normal individuals, but in ischaemic hearts the requirement is reduced.
Respiratory system
Thiopental causes a dose-dependent reduction in both respiratory rate and tidal volume. Manifestations of irritation such as cough and hiccup are uncommon, although it is less effective at depressing the laryngeal reflexes than propofol.
Other effects
Thiopental is not emetic. It has minimal effect on hepatic, renal or adrenal function. Uterine tone is unaffected, but it readily crosses the placenta, also diffusing back into the maternal circulation as plasma levels fall, so this is of little practical significance.
Metabolism
Metabolism occurs in the liver, with an extraction ratio of 0.1–0.4. Phase I mechanisms result in oxidation of the C-5 side chains, replacement of the sulphur with oxygen to produce pentobarbital and cleavage of the barbiturate ring into urea and a three-carbon portion. These actions are robust enough to cope with major liver dysfunction, so thiopental may still be suitable in hepatic failure. Clearance is insufficient to deal with repeated doses, and cumulation therefore occurs.
Steroids
Examples – eltanolone, hydroxydione, minaxolone. At present there are no steroid anaesthetic agents available for clinical use.
Physical
The steroid anaesthetic agents are based on the familiar steroid nucleus. Note that oxy- and hydroxy- substitutions at 3 and 20 appear to be associated with anaesthetic activity.
Clinical
Central nervous system
Most of the steroid agents produce a rapid onset of action (within one arm–brain circulation time) with smooth induction of anaesthesia, and a rapid recovery.
Cardiovascular system
The steroids are less depressant to the cardiovascular system than barbiturates and propofol.
Respiratory system
The steroids are less depressant to tidal volume and respiratory rate than the barbiturates or propofol.
Eltanolone (pregnanolone)
Eltanolone produces rapid induction and recovery of anaesthesia, but this is slower than propofol and accumulation occurs. It is insoluble in water, so a soya bean emulsion is used. It has a terminal half-life of about 50–80 minutes. It has been abandoned because of cutaneous reactions.
Hydroxydione
Hydroxydione is a water-soluble steroid anaesthetic. It has little effect on cardiorespiratory physiology, some neuromuscular blockade with a low incidence of coughing and a very low incidence of nausea and vomiting. However, slow onset of action (several minutes), pain on injection and localised venous irritation have prevented its success.
Minaxolone
Minaxolone is a water-soluble steroid anaesthetic. It has a slow onset of action and its clinical usefulness has been limited by the excitatory activity, associated convulsions and abnormal liver function tests.
Butyrophenones
Examples – benperidol, droperidol, haloperidol
Butyrophenones are covered in detail in Chapter 34.
Phencyclidine derivatives
Physical
Ketamine is a derivative of phencyclidine and cyclohexamine. It is supplied as an aqueous solution in several concentrations, formulated as a weak acid with a pH between 3.5 and 5.5. Ketamine has a single chiral carbon atom. It is supplied as a racemic mixture of the two stereoisomers.
Pharmacodynamics
Ketamine is a non-competitive antagonist of the calcium ion channel operated by the excitatory NMDA glutamate receptor that is likely to be responsible for anaesthesia, analgesia and neurotoxicity. Ketamine also inhibits the NMDA receptor by stereoselectively binding to the phencyclidine (PCP) binding site. Ketamine interacts with the δ, κ and μ opioid receptors. S(+)ketamine provides more potent analgesia than R(–)ketamine. There is stereoselectivity for κ and μ receptors but not δ receptors. Evidence suggests that ketamine may be an antagonist at the μ receptor and that analgesia is not μ-mediated. Ketamine, especially R(–)ketamine, also interacts weakly with the σ receptor (formerly classified as an opioid receptor). The potency of ketamine at the receptors is in the order μ > κ > σ > δ.
High doses of ketamine have a local anaesthetic action, and there is supportive evidence of fast sodium channel blockade.
Ketamine also acts stereoselectively at muscarinic acetylcholine receptors. This action is likely to be antagonist, as ketamine produces anticholinergic effects such as bronchodilatation, delirium and a sympathomimetic action. Ketamine anaesthesia is antagonised by anticholinesterases. It is thought that ketamine may also have an effect on voltage-sensitive Ca2+ channels.
Clinical
Ketamine has a slow onset of action, taking about 1 minute after IV injection to achieve an effect. Ketamine can be administered intramuscularly, with an onset of sleep in 2–5 minutes and duration of 12–25 minutes. The role of extradurally and intrathecally administered ketamine has yet to be established, but, in brief, it causes segmental blockade, and affects the receptors located in the spinal cord as described above.
Central nervous system
Ketamine produces sleep, analgesia and dissociation (a psychological detachment from the surrounding environment). The eyes frequently remain open, and eyelash, corneal and laryngeal reflexes are preserved to a variable extent. Muscle tone is increased and marked involuntary movements are common.
Unlike other typical induction agents, it affects the limbic system rather than the thalamocortical axis. Analgesia is effected by inhibition of the affective, emotional component of pain mediated by the thalamic reticular system rather than the transmission of the nociceptive signals. As well as a slow onset time, the duration of action is much longer than that of the faster induction agents. The recovery may be associated with hallucinations, diplopia or temporary blindness. These effects are a particular problem with short procedures, but those lasting beyond an hour when anaesthesia has been maintained with other agents are less of a problem.
Dissociative side effects are less in males and children, and can be reduced by the concomitant use of opioids, benzodiazepine, droperidol or thiopental. There is no retrograde amnesia.
A significant rise (80%) in CBF, ICP, intraocular pressure (IOP) and CMRO2 may persist for 30 minutes. The onset of effect is associated with a loss of α waves followed by a dominant θ wave on the EEG.
The (+) isomer is more potent than the (–) isomer for hypnosis, analgesia and dissociative effects, supporting the hypothesis that ketamine acts via receptor interaction.
Cardiovascular system
Ketamine increases heart rate, blood pressure and catecholamine levels by a generalised increase in CNS activity. These changes are marked, with 30–100% increases. Direct myocardial depression counteracts the increased sympathetic activity and may leave stroke volume unaffected. Arrhythmias are not common.
Respiratory system
Ketamine has the advantage that, in the absence of opioid, it does not depress respiration greatly. It is a bronchodilator, and protective reflexes are preserved to some extent. Preserved airway maintenance is particularly useful, but reflex protection of the airway from aspiration cannot be guaranteed. Because of the sparing of these reflexes and an increase in secretions, coughing, hiccup and laryngospasm are more prevalent than with thiopental.
Metabolism
The pharmacokinetic profiles of both stereoisomers are identical. Ketamine is metabolised in the liver by N-desmethylation to norketamine. Norketamine has a half to a third of the potency of ketamine and is subsequently hydroxylated. Hydroxylation of the ketamine ring plays a minor role in its metabolism.
Most appears in the bile, with 20% as metabolites in the urine, but some appears unchanged in urine and faeces.
Related posts:






Full access? Get Clinical Tree


