Chapter 84 Hypertension (HTN) is a ubiquitous condition affecting close to one third of Americans and approximately 1 billion people worldwide.1,2 Although tremendous strides have been made over the past 20 years in management of chronic HTN, prevalence has been on a steady rise, and blood pressure (BP) is controlled (defined as below 140/90 mm Hg for most individuals and below 130/80 mm Hg for those with diabetes mellitus or chronic kidney disease [CKD]) in only 50% of patients.3,4 Among those with poorly controlled HTN, over 10% have persistent BP elevations of greater than 160/100 mm Hg—a circumstance that is particularly concerning, because the independent risk of pressure-related cardiovascular mortality is known to double with each 20/10 mm Hg rise in BP above the ‘ ‘ideal’ ’ level of 115/75 mm Hg. Of note, the distribution of HTN is not uniform, and, especially in the United States, individuals of African descent have a higher disease prevalence (41.4% vs. 28.1% for Caucasians) and poorer BP control, leading to an increased risk of adverse outcome.1,4,5 In fact, HTN is the single most important contributor to racial differences in life-years lost from cardiovascular disease, accounting for half of the excess risk within the African American community.6 These data have important implications for the emergency department (ED).7 According to the National Hospital Ambulatory Medical Care Survey, nearly 3.5 million ED visits related to chronic HTN occurred in 2005 (a 75% increase from 10 years earlier); in 2006,8 more than 40% of those treated in EDs across the United States had an initial BP that was moderately elevated (i.e., >140-159/90-99 mm Hg) or severely elevated (i.e., ≥160/100 mm Hg).9 The vast majority of those with increased BP will be asymptomatic (from the perspective of hypertensive end-organ damage), and their BP elevation often will diminish without treatment within a few hours of arrival.10,11 When patients are symptomatic, however, uncontrolled HTN may represent a true emergency that requires rapid intervention. Differentiating patients who warrant immediate treatment for potentially serious complications of acute HTN from those with simple BP elevations with a low likelihood of near-term adverse outcomes is thus a critical aspect of emergency medicine practice. Definition of Hypertension and Relevant Terminology Although BP below 120/80 mm Hg is considered normal, delineation of what constitutes HTN is evolving. For decades, 140/90 mm Hg has existed as the universally established threshold that separates HTN from prehypertension (i.e., the BP range above normal but below elevated) or normotensive state. This cutoff point was derived from studies of manual BP measurements obtained by specifically trained personnel in an office using proper technique in a nonstressful setting.12 Automated BP measurement is more reliable than office-based manual assessment and, when obtained from a patient sitting alone in a quiet environment, yields numerical values that are equivalent to mean BP as measured by the “gold standard”—24-hour ambulatory BP monitoring (ABPM).12–14 Newer automated office BP (AOBP) measurement devices are so much more precise that recent guidelines have suggested lowering the cutoff for establishing a diagnosis of HTN to 135/85 mm Hg (the level above which the American Heart Association considers ABPM measurements abnormal) when BP is obtained via this approach.14,15 The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 7) defines BP levels of 120-139/80-89 mm Hg as pre-HTN, 140-159/90-99 mm Hg as stage I HTN, and 160/100 mm Hg or higher as stage II HTN.3 Despite its inherent usefulness, the JNC 7 classification system lacks sufficient granularity to help emergency physicians discern, even among patients with stage II HTN, specific subgroups for whom intervention in the ED is beneficial. This is especially true for patients with substantial (i.e., ≥180/110 mm Hg) BP elevation, where the focus of care is often erroneously placed on cosmetic improvement in numbers without regard for clinical necessity.16,17 1. Hypertensive emergency: A condition distinguished by acute target-organ dysfunction, manifesting with newly developed clinical sequelae or consequential diagnostic test abnormalities. It has been estimated that 1 to 2% of patients with HTN will experience a hypertensive emergency in their lifetime, and in the United States the frequency of hospital admission for such a diagnosis is approximately 110 per 100,000.18 2. Poorly controlled chronic HTN: A presentation in which patients with established HTN are found to have elevated BP without specific attributable symptoms or evidence of acute target-organ damage. Such presentations most often result from nonadherence to treatment regimens or inadequate medical management but also may reflect refractory disease. Concurrent use of seemingly innocuous medications, including nonsteroidal anti-inflammatory drugs (NSAIDs), steroids, decongestants, appetite suppressants, over-the-counter stimulants, oral contraceptives, and tricyclic antidepressants or rebound from short-acting antihypertensives, such as clonidine, may be contributory. 3. Elevated BP without prior history of HTN: A relatively frequent occurrence in which routine ED vital signs identify an elevated BP. Such individuals also may visit the ED after an outpatient physical examination, community health-screening event, or automated BP measurement by the patient identifies elevated BP. Whether or not this truly represents HTN can be difficult to determine in the ED, and subsequent reexamination in an outpatient setting will usually be required. Studies have shown, however, that approximately 70% of those with high BP in the ED but no established history of HTN will have elevated BP on ABPM or at clinic follow-up.19–21 Patients should not be diagnosed with HTN, however, until a series of properly performed BP measurements in a nonemergency ambulatory setting confirms the diagnosis. Whereas BP is known to rise with increasing age, onset of HTN in the nonelderly represents a complex interplay of multiple inciting factors including neurohormonal dysregulation, vascular modulation, sodium intake, psychosocial stress, and obesity. Alterations in cardiac and renal function are also important, serving as both contributors to and consequences of ongoing BP elevation. Despite an advanced understanding of the pathophysiology of HTN, the definitive cause of elevated BP remains unknown in more than 90% of patients. These individuals are labeled as having primary or essential HTN, and the cause is considered idiopathic. In the subset of patients for whom an identifiable cause can be ascertained, the term secondary HTN applies (Table 84-1). Table 84-1 Secondary Causes of Hypertension The sympathetic nervous system (SNS) has a pivotal role in the development of HTN.22 Norepinephrine, the principal sympathetic neurotransmitter, is a potent stimulator of vasoconstriction. This effect is mediated through peripheral alpha1-adrenergic receptor activation in vascular smooth muscle cells and occurs predominantly in small-diameter arterioles. Although individually these vessels contribute a miniscule amount to BP, in sum they serve as the primary driver of systemic vascular resistance (SVR) and constitute the main force that amplifies afterload in HTN.23 The SNS also stimulates beta1-adrenergic receptors in the heart, leading to an increase in cardiac output (CO) through augmentation of stroke volume and heart rate, but these are considered lesser contributors to the pathologic process of high BP. Sympathoactivation exerts additional, direct effects on the kidney that promote sodium reabsorption leading to an increase in circulating blood volume and trigger renin release, which results in angiotensin II production and further vasoconstriction.24 Independent of SNS activity, the renin-angiotensin-aldosterone system (RAAS) is critical to BP dynamics. Renin is an enzyme produced by juxtaglomerular cells in the kidney in response to several factors, including sodium load in the distal tubule, renal perfusion status, and, as noted earlier, adrenergic stimulation. Renin cleaves angiotensin I from its plasma globulin precursor, angiotensinogen. Angiotensin I is then converted to angiotensin II by circulating and tissue-bound (especially lung) angiotensin-converting enzyme (ACE). Angiotensin II exerts systemic and renal effects by binding to angiotensin II type I (AT1) receptors, which results in arterial vasoconstriction, sodium reabsorption, and modulation of glomerular filtration rate (GFR).25 Through AT1 receptor binding in the adrenal gland, angiotensin II also serves as a potent stimulator of aldosterone release, which promotes further sodium reabsorption and potassium excretion. Continued vascular stimulation by both the SNS and RAAS coupled with an increase in wall tension caused by HTN itself leads to ongoing remodeling throughout the arterial tree.26,27 In large vessels such as the aorta or carotid arteries, this results in increasing intima-media thickness (IMT) with minimal luminal narrowing (unless there is unrelated plaque buildup). In contrast, small-vessel and arteriolar remodeling reduces the lumen diameter.28 Although both forms of remodeling work to normalize wall stress associated with HTN, they reduce vasodilatory capacity and enhance the vasoconstrictor response when faced with a hypertensive stimulus.27 The average American has a daily sodium intake of close to 3500 mg (150 mEq)—more than double the recommended level of 1500 mg (approximately 65 mEq) put forth by the U.S. Department of Agriculture in its 2010 dietary guidelines. Randomized trials have consistently demonstrated a reduction in systolic BP with diminished daily sodium intake (up to 7 mm Hg/1200 mg or 52-mEq decrease in hypertensive individuals).29,30 Salt sensitivity is defined by an increase in BP with intake of a high-sodium diet. It is linked to obesity but may be more directly related to defects in renal ion transport mechanisms that lead to ongoing sodium retention and potassium depletion.31,32 Relative hypokalemia plays a critical role, and the entire effect of salt sensitivity on BP can be abolished with high-dose (approximately 4000 mg or 100 mEq per day) potassium supplementation.5,29 Life stressors, especially socioeconomic status, are known to adversely affect health and wellness.33 There is increasing evidence that such stress is an important modulator of BP (through its effects on SNS function and the hypothalamic-pituitary axis) and a specific contributor to disparities in HTN.34,35 Although episodic stress reactions can lead to transient sympathetic surges, sustained stimulation related to ongoing concern over life circumstances (e.g., financial security, crime and safety, racism) triggers a chronic adaptive response and is emerging as an important consideration in patients with seemingly idiopathic HTN. Increased body mass index (>30 kg/m2) is a known risk factor for HTN (odds ratio 3.4; 95% confidence interval, 2.5-4.6) and explains 20 to 80% of the recent rise in HTN prevalence.36 Elevated BP in obese individuals correlates with high circulating aldosterone and cortisol levels, which in turn may be a related to salt sensitivity. Obesity, especially truncal, is also strongly associated with diabetes and obstructive sleep apnea, both of which contribute to poor BP control. Uninterrupted by treatment, continued vasoconstriction leads to a number of deleterious consequences that culminate in target-organ damage. On a macrocirculatory level, the central component of the cardiovascular system (i.e., heart and large blood vessels) is most affected. Sustained elevations in SVR cause significant augmentation of the pressure wave reflected from the periphery back to the central circulation (termed the augmentation index), thus driving up left ventricular (LV) afterload; the increase manifests with a rise in the central aortic pressure and change in the morphology of its waveform.23,37 This results in increasing impedance to forward flow from the heart, which in turn requires greater contractile force to maintain aortic valve opening and the duration of ventricular ejection.38,39 Active contraction against this resistance also increases intraventricular wall tension, which, together with ongoing stimulation from, among other things, the SNS and RAAS, triggers cardiomyocyte hypertrophy and myocardial fibrosis. Initially, this leads to an increase in LV mass, which enhances the heart’s pumping against excessive afterload. When progressive, however, this process results in LV stiffening and impaired diastolic function with a reduction in LV filling and diminished flow from the left atrium to the LV. If the increase in afterload is sudden, an abrupt decrease in stroke volume occurs, precipitating backflow of fluid into the lungs and rapid onset of “flash pulmonary edema.” If excess afterload is more gradual or even chronic, a subacute rise in LV end-diastolic pressure may cause increased wall tension with compression of the subendocardial microvasculature and non–coronary artery–mediated myocardial ischemia. Over time, this contributes to LV wall thinning, chamber dilation, and eventually systolic dysfunction. Most hypertensive emergencies occur in patients with chronic HTN.40 Organ systems involvement is relatively consistent and is dominated by injury to the brain, heart, or kidneys (Table 84-2). True hypertensive emergencies are defined by the target-organ acutely involved. Focal neurologic deficit or altered mentation therefore point to brain injury, whereas chest pain or shortness of breath is indicative of cardiac or vascular involvement. Although frequently accompanied by an elevated BP, symptoms such as headache, epistaxis, and dizziness are not, in and of themselves, evidentiary of end-organ injury and in isolation, do not constitute a hypertensive emergency, nor indicate the need for acute BP reduction. Table 84-2 Hypertensive Emergencies by Organ System *Range based on percentages compiled from Szczech and others,106 Katz and others,52 and Zampaglione and others.111 Adapted from Levy P: Hypertensive Emergencies: On the Cutting Edge. Advancing the Standard of Care: Cardiovascular and Neurovascular Emergencies. EMCREG-International Symposia Monograph, February 2011, pp 19-26. Available at www.emcreg.org. Acute stroke per se is not a hypertensive emergency, although treatment of acute intracranial hemorrhage often includes BP control (see Chapter 101). Chronic HTN is the primary population-attributable risk factor for development of heart failure,41 and more than 50% of ED patients with acute cardiac decompensation have HTN (see Chapter 81). Although the relationship between acute systemic HTN and pulmonary edema is not uniform, in up to half of cases, HTN may be the sole cause or a major contributor to the acute episode.42,43 Acute heart failure often responds well to vasodilatory agents and a reduction in afterload. Similarly, acute BP management may play a key role in therapy of acute coronary syndrome and demand ischemia (see Chapter 78), and acute aortic dissection (see Chapter 85). Acute kidney injury in the setting of elevated BP may be a consequence of associated target-organ damage, especially acute heart failure, and particularly when such patients are on baseline diuretic or calcium channel blocker therapy. Recent or chronic NSAID or newly initiated ACE inhibitor (ACEI) therapy may also contribute, but the effect of these agents are usually transient (see Chapter 97).44 Preeclampsia and eclampsia are discussed in Chapter 178. Most patients who are found to be significantly hypertensive on intake vital sign measurement or who come to the ED because BP was found to be elevated in an outpatient setting or by self-measurement do not have an acute hypertensive emergency, and for such patients, acute reduction of BP is not indicated. These patients do not have target-organ injury and most often have poorly controlled chronic HTN, HTN in the context of nonadherence to medication regimen, or, less commonly, elevated BP without prior history of HTN. When symptoms are present, they generally are nonspecific (e.g., low-grade or recurrent headache, atypical chest pain, dyspnea, dizziness, generalized weakness, focal but anatomically uncorrelated weakness or numbness, vague visual disturbances) and, with the exception of dyspnea, appear to be unrelated to the degree of BP elevation.45,46 Despite widespread belief among the public (and some physicians) to the contrary, existing evidence does not support a causal relationship between epistaxis and acute, severe HTN.47,48 There is no evidence of benefit for acute BP reduction in patients with these vague symptoms and elevated BP, regardless of the level. If chronic medications have been missed, as usually is the case, these should be restarted. The patient should be observed in a quiet area of the ED and reassured; in many cases the BP spontaneously improves.10 There are no data supporting either a threshold BP warranting treatment or a target BP to be achieved before discharge. Although well-intentioned, some misguided providers may be tempted to administer a short-acting, potent antihypertensive agent such as clonidine or hydralazine to achieve an immediate but transient (and clinically meaningless) reduction in BP. This practice is without rationale or evidence of benefit and, according to a recent retrospective cohort study, may be associated with an increased likelihood of subsequent ED visitation for issues related to HTN.49 Funduscopy can provide evidence of hypertensive target-organ damage. Findings of acute hypertensive retinopathy include focal intraretinal periarteriolar transudates (whitish ovoid lesions deep in the retina), focal retinal pigment epithelial lesions (evidence of choroidal injury), macular and optic disk edema, and cotton-wool spots (fluffy white lesions that consist of swollen, ischemic axons caused by small-vessel occlusion).50 Hard exudates, which consist of lipid deposits located deep in the retina, are also a common but late occurrence. When identified, such funduscopic abnormalities are considered diagnostic; however, they may be absent in more than 30% of patients with a clinically evident hypertensive emergency.51 Lesions of acute retinopathy are distinct from more chronic changes, which include arterial narrowing, “copper” or “silver” wiring of the arterioles, arteriovenous nicking, and retinal hemorrhages. The spectrum of retinal findings in HTN can be graded on a 5-point scale (Box 84-1). Despite its potential usefulness, funduscopy is infrequently and inadequately performed in the evaluation of severely elevated BP in the busy ED.45,52
Hypertension
Perspective
Principles of Disease
Pathophysiology of Chronic Hypertension
CAUSE
DIAGNOSTIC TEST
CLINICAL CLUES
Endocrine
Cushing’s syndrome and other glucocorticoid excess states
History; dexamethasone suppression test
Glucose intolerance; purple striae
Hyperaldosteronism and other mineralocorticoid excess states
24-Hour urinary aldosterone level or other mineralocorticoids
Unexplained hypokalemia
Oral contraceptive use
History
Pheochromocytoma
24-Hour urinary metanephrine and normetanephrine
Labile or paroxysmal HTN with palpitations, pallor and perspiration
Thyroid disease
Parathyroid disease
Serum TSH
Serum PTH
Temperature intolerance, weight loss, tachycardia
Hypercalcemia
Pulmonary
Obstructive sleep apnea
Sleep study with O2 saturation
Obesity; narcolepsy
Renal
Chronic pyelonephritis
History; urinalysis, urine culture
Diabetic nephropathy and other chronic kidney disease
Estimated GFR; urine albumin/creatinine ratio
Nephritic and nephrotic syndromes
Urinalysis
Polycystic kidney disease
Renal ultrasound
Renovascular conditions (renal artery stenosis)
Doppler flow study; magnetic resonance angiography
HTN onset before the age of 30 yr or after 55 yr; abdominal bruit; refractory HTN control; recurrent pulmonary edema; unexplained renal failure
Toxic or Metabolic
Chronic alcohol abuse
History; ETOH level
Sympathomimetic drug use
History; drug screen
Tyramine containing foods
History
Paroxysms of HTN, especially in those taking monoamine oxidase inhibitors
Vascular
Atherosclerosis
Coarctation of the aorta
CT angiography
Decreased lower extremity pulses
Neurohormonal Dysregulation
Vascular Modulation
Sodium Intake
Psychosocial Stress
Obesity
Pathophysiology of Target-Organ Damage
Clinical Features
Hypertensive Emergency
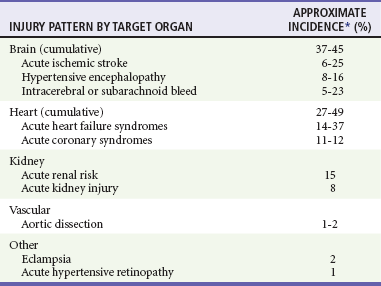
Acute Stroke, Heart Failure Syndromes, and Other Hypertension-Related Emergencies
Blood Pressure Elevation in the Absence of Target-Organ Dysfunction
Diagnostic Strategies

Full access? Get Clinical Tree


Anesthesia Key
Fastest Anesthesia & Intensive Care & Emergency Medicine Insight Engine




