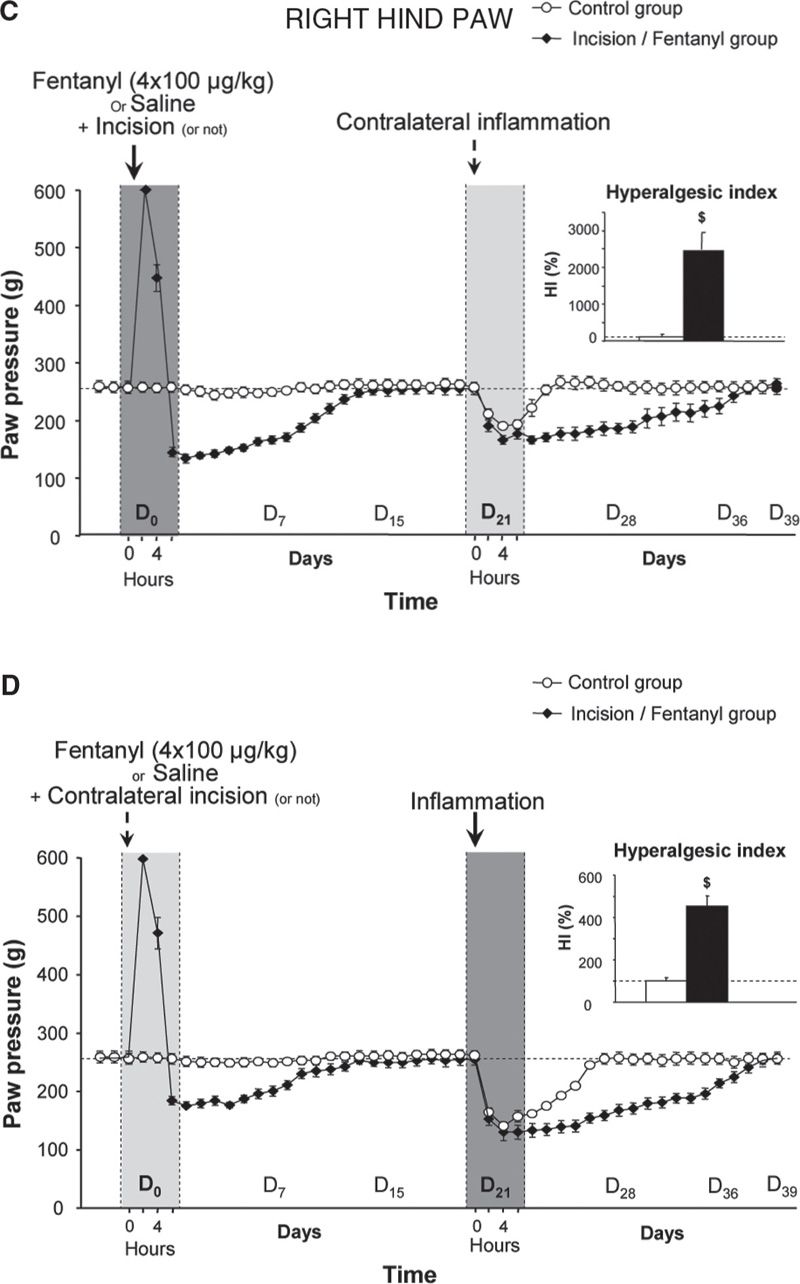
FIGURE 1 Latent pain hypersensitivity induced by a surgical lesion and its amplification by the administration of opioid analgesics. The mechanical nociceptive threshold (vocalization elicited in response to pressure) in rats was assessed using a Randall-Selitto test on the posterior paws. On D0, the rats received four injections of saline or fentanyl (100 µg/kg) at intervals of 15 minutes, and an incision was made in the plantar muscle of the left paw five minutes after the first injection of fentanyl or saline. Incision was not performed on control animals. Three weeks later (D21), an injection of carrageenan, which causes inflammation, was administered to the posterior right paw. The new inflammatory lesion created on D21 induced an exaggerated pain response (hyperalgesia) in the inflamed paw (B and D – right hind paw), but was capable of reactivating the pain hypersensitivity in the healed paw, in which there was a surgical lesion, regardless of whether the animals received fentanyl treatment 3 weeks prior (A and C – left hind paw). (Adapted from Laboureyras E, Chateauraynaud J, Richebe P, Simonnet G. Long-term pain vulnerability after surgery in rats: prevention by nefopam, an analgesic with antihyperalgesic properties. Anesth Analg 2009;109:623–31).
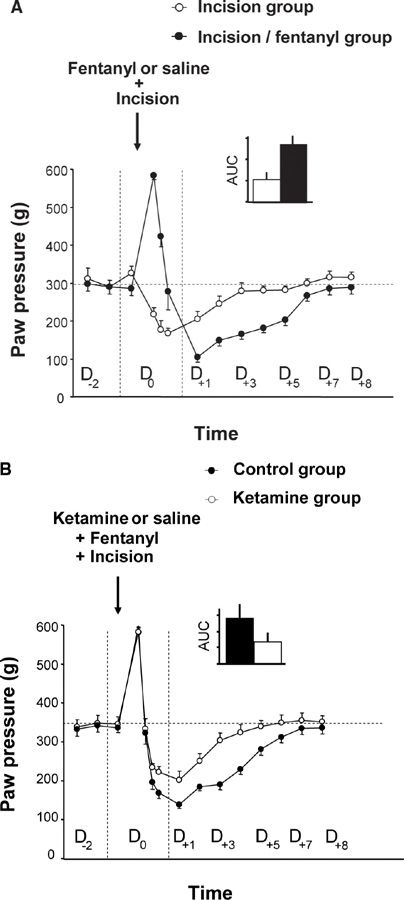
FIGURE 2 Amplification of surgical injury-induced hyperalgesia via the administration of an opioid analgesic and the reduction of this amplification by the administration of ketamine, an NMDA receptor antagonist. (A) On D0, the rats received four injections of saline or fentanyl (100 µg/kg) at intervals of 15 minutes and an incision was made in the plantar muscle of the left paw 15 minutes after the first injection of fentanyl or saline. The administration of fentanyl resulted in an immediate analgesic effect but also potentiated the pain hypersensitivity induced by a surgical lesion, which is expressed as an amplification of the post-surgical hyperalgesia for a several days. (B) Using the same experimental design, three injections of ketamine (3 x 10 mg/kg) or saline were administered subcutaneously. The first injection was administered 30 min before surgery, and the subsequent injections were administered every 5 h. The administration of ketamine results in a reduction of hyperalgesia induced by a surgical lesion associated with fentanyl administration. (From Richebe P, Rivat C, Laulin JP, et al. Ketamine improves the management of exaggerated postoperative pain observed in perioperative fentanyl-treated rats. Anesthesiology 2005;102:421–8).
From a mechanistic standpoint, we and others have demonstrated the critical role of NMDA-Rs in OIH by showing that a single administration of an NMDA-R antagonist, such as ketamine, MK-801 or BN2572 completely prevented OIH in animals [7,8,27,29]. Electrophysiological studies have demonstrated that µ-opioid receptor (MOR) stimulation activates intracellular protein kinase C (PKC) which reduces Mg2+ blocking, thereby activating NMDA-Rs [10]. The subsequent increase in the intracellular Ca2+ concentration has been suggested to further stimulate PKC activity, leading to a lasting enhancement of glutamatergic synaptic efficiency in a positive feedback loop (Fig. 3), a process resembling LTP. Recently, it has been proposed that opioid-induced LTP in the spinal cord results from increased presynaptic glutamate release from the transient receptor potential vanilloid 1 (TRPV1)-expressing primary afferents [56]. Activation of TRPV1 has been reported to contribute to OIH via glutamate release [54], and TRPV1 blockers reduce OIH [11]. Interestingly, blocking of the NMDA-NO cascade using the nitric oxide synthase inhibitor NG-nitro-L-arginine methyl ester (L-NAME) has been reported to produce effects similar to those of NMDA-R antagonists. This result suggests a critical role for this intracellular secondary messenger system in OIH because NO release facilitates the glutamate release at the spinal cord level; this is also consistent with the observation that OIH is significantly attenuated in mutant mice lacking inducible nitric oxide synthase (iNOS) [6]. Taken together, these results indicate that the spinal NMDA-Rs/iNOS system plays a critical role at the dorsal horn level in OIH. Other mechanisms underlying OIH could not be excluded at either the spinal or supra-spinal level, such as the involvement of the β-adrenergic receptor [13], alpha 2-adrenoreceptors [39], Ca2+/calmodulin-dependent protein kinase IIα [12] and local cytokine production [32]. Ca2+/calmodulin-dependent protein kinase IIα (CAMKII) has been described to interact with the NMDA-R system [24]. Therefore, CAMKII and the NMDA receptor systems may interact in a feed-forward manner in OIH. Anti-opioid peptides (AOPs), such as cholecystokinin (CCK), neuropeptide FF (NPFF), dynorphin and orphanin FQ/nociceptin, are endogenous peptides released following opioid receptor stimulation. AOPs have been proposed to be involved in OIH because specific AOP receptor antagonists prevent OIH [47]. AOPs promote the release of glutamate, and AOPs release is facilitated by NMDA-R stimulation [23,48]. Finally, numerous studies have indicated that the rostral ventromedial medulla (RVM) and the surrounding tissue are a prominent source of descending pain modulation (see Descending Modulatory systems by Mary M Heinricher, Part II, Chapter 8 in this book).
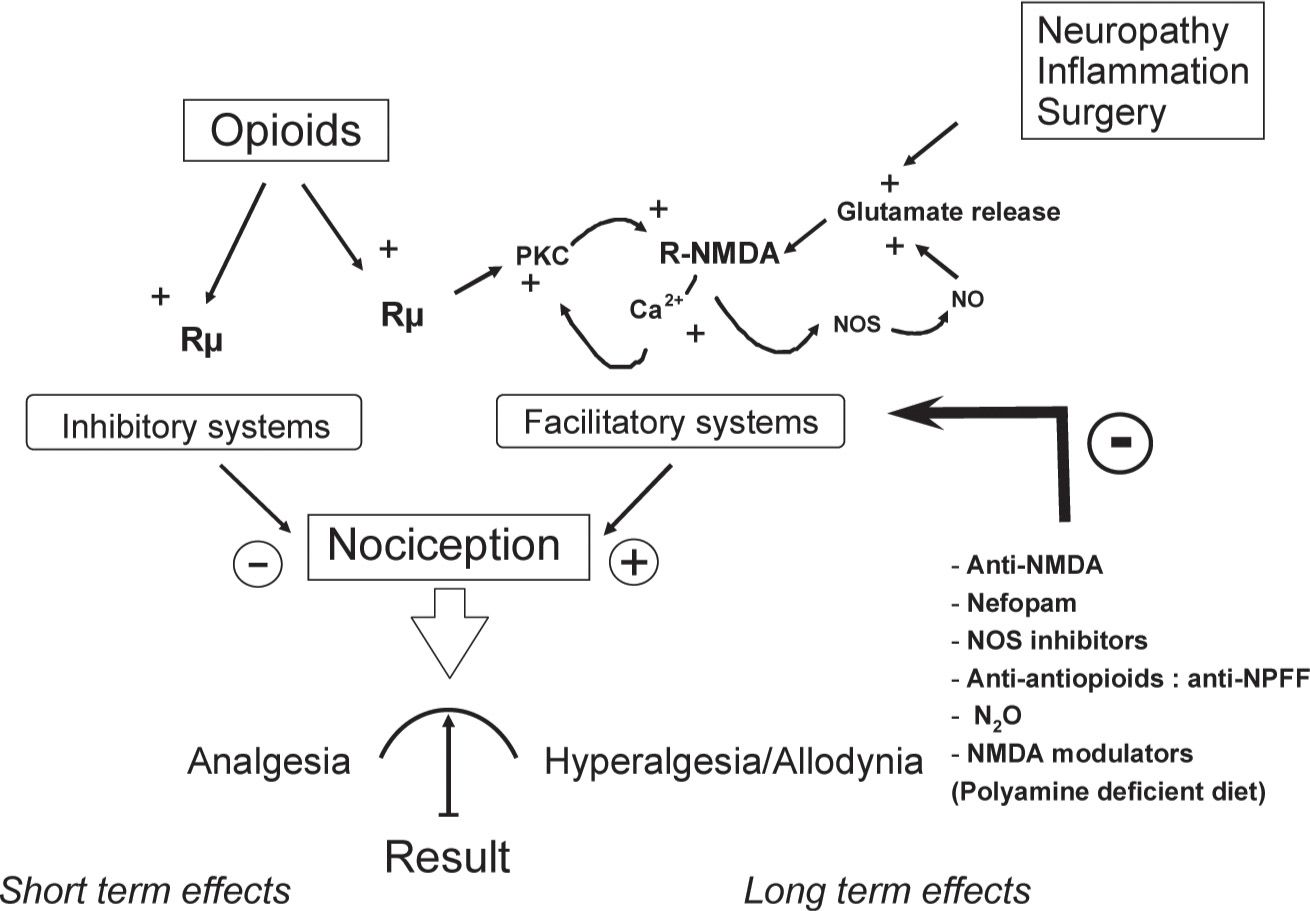
FIGURE 3 Model of the dual effects of opioids. Opioids simultaneously activate pain inhibitory systems (analgesia) and N-methyl-D-aspartate (NMDA)-dependent pain facilitatory systems, inducing pain hypersensitivity that outlasts opioid analgesia. The analgesic effect of opioid administration results from these two opposing processes, suggesting that the effect is partially masked as early as the first opioid exposure and that it is followed by hyperalgesia/allodynia. Repeated opioid administration induces lasting enhancement of glutamate synaptic efficiency in a positive feedback loop, increasing the activation level of NMDA-dependent pain facilitatory systems (sensitization): however, opioid analgesia remains unchanged. This effect leads to an apparent decrease in opioid analgesic potency (apparent tolerance).
From an adaptive viewpoint, OIH, which involves experiencing paradoxical or abnormal pain, may be considered a physiological or normal response if opioid-induced analgesia is considered a disturbance of homeostatic equilibrium. In this context, pain hypersensitivity appears to be an effective response through which a new homeostatic equilibrium can be achieved (i.e., allostasis). The concept of a perturbed equilibrium, described by Canon, exists alongside the concept of physiological balance: the imbalance that has been induced (analgesia) must be compensated for a return to a new equilibrium via the activation of an opponent process [50], such as pronociceptive system recruitment. Similar pain hypersensitivity has been observed following administration of triptans and α2-adrenoreceptor agonists [16,39].
THE ADMINISTRATION OF EXOGENOUS OPIOIDS AMPLIFIES THE LONG-LASTING HYPERALGESIA INDUCED BY A SURGICAL LESION
Considering these new physiological concepts, we must evaluate whether the treatment with an opioid during the perioperative period is likely to amplify post-surgical pain and to promote the development of chronic post-surgical pain. This question is difficult to address in clinical practice because it is difficult to differentiate between normal and abnormal post-surgical pain. To mimic such a surgical situation, we used the preclinical incisional pain model (unilateral incision of a hind paw) developed by Brennan and colleagues [4], in association with the administration of fentanyl administration, an opioid drug that is extensively used by anesthesiologists.
In the first series of experiments, we evaluated the putative enhancement of post-surgical pain in rats receiving a series of 4 subcutaneous fentanyl boluses within 1 hour. Pain scoring was performed by measuring changes in the nociceptive threshold (heat and mechanical tests) or by evaluating changes in postural disequilibrium (weight bearing test) before, during and several days after surgery. These preclinical studies showed that the administration of fentanyl not only produces an immediate analgesia, as expected, but also acts in a dose-dependent manner to strongly potentiate the pain hypersensitivity induced by the surgical lesion [26,41] (Fig. 1C, D). These results have been reproduced in the mouse model by other groups after injections of fentanyl, remifentanil, alfentanil or sufentanil [5,9,37].
As in OIH, this process of sensitization to pain is NMDA-dependent and can be prevented by a single injection of an NMDA-R antagonist, such as ketamine or other medications that are classified as anti-hyperalgesics (nitrous oxide [3], nefopam [26] or gabapentin [53]). The amplification of post-surgical pain was considerably reduced in mice lacking the iNOS gene, confirming the critical role of the NMDA-Rs/iNOS system. Interestingly, this pain hypersensitivity is not only localized to the injured hind paw but is also present on the contralateral uninjured hindpaw, which confirms the central origin of this process [26,43].
It is questionable whether opioids enhance postsurgical pain in humans. Among various clinical studies, only one quantitative sensory test (QST) has clinically demonstrating postsurgical pain enhancement via perioperative opioid use [21]. This study revealed that a relatively large dose of intraoperative remifentanil (0.40 µg kg-1. min-1) increases pain sensitivity, as evidenced by a reduction of the postsurgical pain threshold proximal to the surgical wound and an extension of peri-incisional hyperalgesia. This phenomenon lasted throughout the postoperative period and was associated with an increase in the postoperative self-administration of morphine (patient-controlled analgesia or PCA). In agreement with preclinical observations, a small dose of ketamine during and after surgery prevented the increase in postsurgical pain sensitivity and punctate hyperalgesia and reduced the morphine requirement by PCA. However, human studies have led to conflicting results, most likely because the potential existence of such a phenomenon is framed within the current understanding of opioids as analgesic drugs, rather than hyperalgesic drugs. Moreover, physicians are reticent to undertreat pain and the idea that potent analgesic such as opioids may actually worsen pain symptoms has appears paradoxical and needs better incorporated into our philosophy of pain management. The suggestion that perioperative opioids may actually worsen pain presents new challenges. As it has been suggested [55], the demonstration of such a phenomenon requires rigorous research protocols for serial determination of stimulus dose-response curves under standardized conditions, such as QST, as it has been performed for OIH in human volunteers.
In a second series of experiments, we evaluated the level of long-term pain vulnerability induced by surgery associated with fentanyl by testing the pain response induced by an experimental inflammation performed 3 weeks after surgery. Indeed, it is unclear how long the change of state, in terms of sensitivity to pain, persists beyond the early postoperative period of hyperalgesia induced by tissue injury, even in a latent form when nociceptive inputs have stopped. This question is clinically important because some patients must undergo treatments during the postsurgical period that are potentially painful, while others must undergo further surgeries in the short- or medium-term that are sometimes performed on the contralateral side, such as surgery for hallux valgus, hip prosthesis or cataracts.
In our experimental model, we observed [26] that the amplitude and duration of the hyperalgesia induced by an experimental inflammation in a hind paw (e.g., the right hind paw) was dramatically enhanced when it was performed in rats that had experienced a prior tissue injury, such as a surgical lesion on the contralateral side (e.g., on the left hind paw) (Fig. 1B). This phenomenon, which is referred to as latent pain sensitization, was strongly amplified in the animals that received fentanyl boluses after a surgical incision was performed 3 weeks earlier (Fig. 1D). Similar results were obtained in the same mouse model after perioperative remifentanil administration [5]. These preclinical studies indicate that the administration of an opioid analgesic amplified the development of a latent pain sensitization: despite tissue healing, the individual did not return to the initial physiological state (homeostatic state), but rather developed long-lasting pain vulnerability (allostatic state). This latent pain vulnerability has not yet been explored in surgical patients and must be examined. These data suggest that the evaluation of individual pain and opioid histories in humans may help identify patients at risk for exaggerated postsurgical pain, which may result in chronic post-surgical pain.
ENDOGENOUS OPIOIDS RELEASED DURING ENVIRONMENTAL STRESS ALSO INDUCE PAIN VULNERABILITY
Related posts:
 Neuroimaging and Post-Operative Persistent Pain
Neurotransmitters, Neuromodulators, and Ionophores in Mechanisms of Pain Persistence after Surgery: From Periphery to the Brain
Neuroimaging and Post-Operative Persistent Pain
Neurotransmitters, Neuromodulators, and Ionophores in Mechanisms of Pain Persistence after Surgery: From Periphery to the Brain
 Electrophysiological Techniques: Assessment of Spinal Nociceptive Processing Using the Withdrawal Reflex
Electrophysiological Techniques: Assessment of Spinal Nociceptive Processing Using the Withdrawal Reflex
 Mechanistic Quantitative Sensory Testing: From Animals to Patients
Mechanistic Quantitative Sensory Testing: From Animals to Patients
 Electrophysiological Techniques to Study the Supraspinal Responses to Nociceptive Input in Humans
Electrophysiological Techniques to Study the Supraspinal Responses to Nociceptive Input in Humans
 Drug Effects and Altered Perioperative Pain Processing
Drug Effects and Altered Perioperative Pain Processing

Full access? Get Clinical Tree


