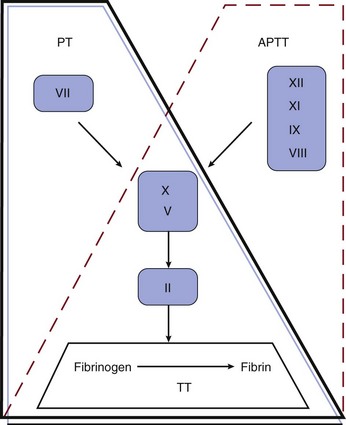
78 APPROACH TO A CRITICALLY ILL PATIENT WITH HEMORRHAGE OR THROMBOSIS LABORATORY TESTS OF COAGULATION COMPLEX THROMBOHEMORRHAGIC DISORDERS CATASTROPHIC ANTIPHOSPHOLIPID SYNDROME Although not reflective of the current model of coagulation, the integrity of coagulation is routinely tested by a limited set of in vitro assays (Fig. 78.1). The prothrombin time (PT) reflects the cascade of reactions traditionally called the extrinsic pathway, whereas the activated partial thromboplastin time (aPTT) reflects the intrinsic pathway. They intersect in the common pathway. The thrombin time (TT) measures the rate of conversion of fibrinogen to insoluble fibrin polymer after thrombin is added to plasma. A prolonged TT may be due to an inhibitor of thrombin (e.g., heparin, direct thrombin inhibitor [DTI]), hypofibrinogenemia or dysfibrinogenemia, fibrin degradation products (FDPs), and rarely paraproteins. The only specific coagulation factor that is routinely measured is fibrinogen. These four tests can usually localize abnormalities in the coagulation cascade. Thrombocytopenia is common in the intensive care unit (ICU). It has been estimated that 23% to 41% of patients in the ICU have a platelet count less than 100,000/µL, and 10% to 17% have a count less than 5000/µL.1 Common causes of thrombocytopenia in the ICU are shown in Box 78.1. In complex, acutely ill patients, many of these mechanisms may operate simultaneously. Severe sepsis is the most common cause of thrombocytopenia in the ICU.2 Frequently, thrombocytopenia must be managed without a specific diagnosis. Medications should be reviewed for potential offending agents.3,4 Inhibitors of platelet function should be avoided with platelet counts below 50,000/µL. If there is bleeding or if invasive procedures are anticipated, platelet transfusions should be given, unless contraindicated, to elevate the platelet count above 50,000/µL. In life-threatening situations, the goal should be a platelet count higher than 100,000/µL. In nonbleeding patients, maintenance of a platelet count above 10,000/µL (20,000/µL with fever or infection) with prophylactic transfusions is usually adequate.5 Pseudothrombocytopenia is a laboratory artifact.6 The platelet count is factitiously lowered because of the presence of naturally occurring antibodies that cause platelet agglutination in the presence of ethylenediaminetetraacetic acid (EDTA) at room temperature. The diagnosis is suspected by finding platelet clumps on the peripheral blood smear. Repeating the platelet count with a different anticoagulant such as citrate will generally produce a normal platelet count. A limited number of drugs have evidence-based data to support a causal role in the development of thrombocytopenia.3,4,7,8 Medications commonly used in the ICU can cause thrombocytopenia (Box 78.2). Drug-induced thrombocytopenia most commonly occurs 7 to 21 days after exposure to the offending agent. Clinical manifestations can range from an asymptomatic decrease in platelets to life-threatening bleeding. The diagnosis is established by (1) finding a temporal relationship between starting the drug and the fall in the platelet count, (2) having no alternative diagnosis, and (3) having the platelet count recover after removal of the putative offending agent. Unfortunately, this is usually difficult to establish in the typical ICU patient. Treatment is based on removing the putative offending agent and initiating a drug of another class if possible. Though often used, steroids in general have not been shown to hasten the rate of platelet recovery. In severe thrombocytopenia with bleeding such as seen with quinine, intravenous immunoglobulin (IVIG) (1 g/kg/day for 2 days) and platelet transfusion are beneficial. All platelet glycoprotein (GP) IIb/IIIa inhibitors have been associated with severe thrombocytopenia that can occur within hours of exposure (up to 2 weeks with abciximab).7 Heparin-induced thrombocytopenia (HIT) is the main differential diagnosis. Bleeding is very uncommon with HIT because of the strong prothrombotic state. Conversely, with GP IIb/IIIa inhibitor-associated thrombocytopenia, bleeding or hematoma formation may occur, especially at the site of the sheath. A platelet count less than 20,000/µL and clinical bleeding are indications for platelet transfusion. The use of IVIG and corticosteroids is not evidence based. Guidelines for the diagnosis and management of ITP have been developed.9,10 Indications for therapy in ITP are platelet count less than 20,000 to 30,000/µL or clinical bleeding. Corticosteroids (prednisone, 1 mg/kg/day, or pulse dexamethasone, 40 mg/day for 4 days monthly) are the usual initial therapy for ITP.9,10 Individuals with ITP in the ICU usually have severe or life-threatening bleeding. Several modalities of therapy should be used in concert to raise the platelet count in urgent situations (methylprednisolone, 1 g/day for 3 days, and IVIG, 1 g/kg/day for 2 days). Although platelets may be destroyed quickly, platelet transfusions should still be used as initial therapy. The response to platelet transfusion may improve after IVIG is given. Anti-Rh(D) IgG (WinRho), 50 to 75 µg/kg, has also been used.11 Because the dose of anti-Rh(D) must be reduced in face of anemia, its use may be problematic in a patient with severe bleeding. Anti-Rh(D) IgG is ineffective in Rh-negative patients and after splenectomy. ε-Aminocaproic acid (4-5 g IV followed by either 2-4 g IV every 4 hours or 0.5-1.0 g/hour continuous IV infusion [maximum 24 g/24 hours]) may be useful for mucosal bleeding and severe menorrhagia. Post-transfusion purpura (PTP) is a rare condition characterized by acute, severe immune-mediated thrombocytopenia. PTP occurs in human platelet antigen-1 (HPA-1) negative individuals who receive HPA-1 positive platelets (most commonly as a contaminant in packed red blood cells [RBCs]). These individuals have previously been sensitized to HPA-1 through transfusion or pregnancy. Usually 7 to 10 days after reexposure to HPA-1, an anamnestic response occurs, resulting in a precipitous fall in the platelet count. Petechiae and bleeding are common. IVIG and plasma exchange are effective.12 Aspirin irreversibly acetylates cyclooxygenase (COX), inhibiting platelet function for the life of the platelet (7 to 10 days). The aspirin effect can be overcome with platelet transfusion or the infusion of desmopressin (DDAVP). The effect of NSAIDs is reversible and disappears as the drug is cleared, usually within 24 to 48 hours for ibuprofen. The risk of bleeding from NSAIDs is lowest with ibuprofen and greatest with ketorolac. The thienopyridine clopidogrel tightly binds the platelet ADP P2Y12 receptor. Clopidogrel should be withheld for 5 to 7 days before elective surgery or invasive procedures.13 In an emergency, platelet transfusion can be tried, but platelet function may not be fully restored because of circulating active clopidogrel metabolites, which have a half-life of 8 hours and bind to the transfused platelets. Prasugrel is a new thienopyridine whose active metabolites have a 4-hour half-life. COX-2 inhibitors don’t affect platelet function. The hemorrhagic diathesis of renal failure is the result of metabolic derangements related to uremic toxins.14 Bleeding may worsen when the hematocrit falls below 30% due to a rheologic phenomenon in which rapidly flowing RBCs gravitate to the center of the streaming blood and force the platelets toward the vessel wall. Uremic bleeding is uncommon in the modern era of dialysis.15 For clinical bleeding, intravenous DDAVP (0.3 µg/kg given over 30 minutes) is often therapeutic.14 If the hematocrit is less than 30%, the patient should be transfused with packed RBCs.14,15 An alternative to DDAVP is cryoprecipitate (10 bags every 12 to 24 hours).14 Intravenous conjugated estrogen, 0.6 mg/kg/day for 5 days, is also effective.14 An unexpected “coagulopathy” (prolonged aPTT and TT) may develop as a result of delayed clearance of heparin after dialysis. If available, an anti–factor Xa assay will show the presence of heparin. Although commonly looked for, HIT is an uncommon cause of thrombocytopenia in the ICU.2,16 HIT is a paradoxical condition in which modest thrombocytopenia may be associated with devastating heparin-induced thrombocytopenia thrombosis (HITT). Thus, the entire health care team needs to be vigilant for the development of HIT in any patient receiving heparin. HIT has been associated with all types of heparin (unfractionated [UFH] and low-molecular-weight [LMWH]), at any dose, and by any route, including flushes and heparin-coated catheters.1,17 The incidence of HIT has been estimated to be less than 1% of patients in the ICU.2,16,18 HIT occurs in three time intervals. Classic HIT occurs 5 to 15 days after the initiation of heparin. Rapid-onset HIT develops hours to 1 to 2 days after heparin is started in individuals who have preformed circulating antibodies from a previous exposure to heparin, usually in the last 2 months. Classic and rapid-onset HIT may be manifested as thrombocytopenia with or without thrombosis [HIT(T)]. Delayed-onset HIT occurs an average of 12 days after the discontinuation of heparin and is manifested as isolated thrombosis. The thrombocytopenia of HIT is usually modest with an average platelet count of 50,000 to 60,000/µL.19 Severe thrombocytopenia (<10,000/µL) should suggest an alternative diagnosis. HIT may be associated with a normal platelet count if there is a baseline thrombocytosis (e.g., postoperatively). HIT may be manifested as isolated thrombocytopenia or thrombocytopenia with potentially devastating thrombosis. Venous complications include deep venous thrombosis (DVT), pulmonary embolism (PE), cerebral sinus thrombosis, infarctive adrenal hemorrhage, and skin necrosis at the heparin injection site. Arterial complications include iliofemoral artery thrombosis, digital ischemia, myocardial infarction, stroke, and mesenteric artery thrombosis. HIT is an immune-mediated process in which the heparin–platelet factor 4 complex becomes immunogenic. Antibodies are formed that activate platelets, causing the release of thrombogenic microparticles.16,19 The antibodies can also activate monocytes and endothelial cells. The diagnosis of HIT is clinicopathologic. HIT should be thought of in appropriate clinical situations (Box 78.3). The “4T” score has been used to predict the likelihood (pretest probability) of HIT (Table 78.1).16,20 The HEP score, devised by an expert consensus panel, has recently been validated.21 Although the decision to initiate therapy should be based on the clinical likelihood of HIT, laboratory testing should be used for the retrospective confirmation of the pretest likelihood of HIT. The complexities of HIT antibody testing are reviewed elsewhere.16,22 In general, a strongly positive HIT enzyme-linked immunosorbent assay (ELISA) and strongly positive serotonin release assay (SRA) with a high pretest probability confirm the diagnosis of HIT, whereas a negative ELISA and SRA make the diagnosis of HIT unlikely with a low pretest probability. In other situations, clinical judgment prevails in establishing or excluding a diagnosis of HIT, especially because ELISA and SRA testing from commercial labs has not been validated in clinical studies.1,16,22 Table 78.1 Determining Pretest Probability of Heparin-Induced Thrombocytopenia (HIT): The “4 Ts” *First day of heparin exposure = day 0. Adapted from Lo GK, Juhl D, Warkentin TE, et al: Evaluation of pretest clinical score (4 T’s) for the diagnosis of heparin-induced thrombocytopenia in two clinical settings. J Thromb Haemost 2006:4:759. Once the diagnosis of HIT is thought to be likely, all heparin should be discontinued. Heparin-coated pulmonary artery catheters should be replaced with noncoated catheters. Catheters for dialysis or apheresis should be locked with 4% citrate or tissue plasminogen activator. The patient’s chart and bedside should be labeled heparin allergy. Platelet transfusions should be avoided except for life-threatening bleeding, given the anecdotal observations of acute thrombosis occurring after platelet transfusion. A patient with life- or limb-threatening arterial thrombosis should be evaluated for surgical intervention. Anticoagulation with a DTI (Table 78.2) should be initiated in all patients unless contraindicated. LMWH is contraindicated in HIT caused by UFH. Conversion to warfarin can be considered after a minimum of 5 days of DTI therapy if the platelet count has returned to normal (suggesting that the process has cooled off and the patient is no longer hyperprothrombotic) and no future invasive procedures are planned. Simply discontinuing heparin therapy and not starting a DTI is inappropriate because occult thrombosis may have already developed.23 Starting or continuing warfarin monotherapy is also contraindicated because of the risk of venous limb gangrene.24 If warfarin has been given at the time HIT is diagnosed, vitamin K should be given to replenish proteins C and S. If invasive procedures are needed (e.g., tracheostomy, pacemaker), it is best to delay them, if medically safe, until the platelet count is normal to minimize the risk of developing thrombosis during the time that DTI therapy is withheld. Inferior vena cava (IVC) filters should be avoided because of the risk of vena cava thrombosis. Patients with active HIT may need percutaneous coronary intervention (PCI). Argatroban has been approved by the Food and Drug Administration (FDA) for use during PCI in patients with HIT.25 Though not FDA approved, bivalirudin has been used safely in this situation.26 In a patient who has active HIT or persistent HIT antibodies and who needs open heart surgery, medical management is recommended until the antibody becomes negative. If urgent surgery is needed, bivalirudin is most commonly used.27 For the patient with a past history of HIT who needs open heart surgery and is currently HIT antibody negative, heparin can be used during bypass and a DTI started as soon as it is surgically safe postoperatively.28 Table 78.2 Parenteral Anticoagulant Doses *Suggested dose modification in ICU patients. Data from Selleng K, Warkentin TE, Greinacher A. Heparin-induced thrombocytopenia in intensive care patients. Crit Care Med 2007;35:1165-1176. Thrombotic thrombocytopenic purpura (TTP) is a relatively rare disorder whose hallmarks are thrombocytopenia, microangiopathic hemolytic anemia (MAHA), and neurologic dysfunction. The diagnosis of TTP should be considered in any patient with unexplained thrombocytopenia and MAHA.29 TTP is the clinical manifestation of a heterogeneous group of underlying disorders driven by different pathophysiologic processes. Although most cases of TTP are idiopathic, TTP may be associated with exposure to drugs (e.g., ticlopidine, clopidogrel, quinine, mitomycin C, gemcitabine, cyclosporine), pregnancy, HIV infection, and bone marrow transplantation.30 The morphologic hallmark of TTP is hyaline thrombi in precapillary arterioles composed primarily of platelets. Classic TTP is due to an immune-mediated deficiency of the metalloproteinase ADAMTS13, which cleaves ultralarge multimers of von Willebrand factor (vWF) into smaller forms that are less platelet reactive.31 Deficiency of this enzyme allows ultralarge forms of vWF that are normally sequestered in the endothelium to bind platelets and form microthrombi in the circulation. Conversely, ADAMTS13 activity is normal in TTP associated with bone marrow transplantation and the hemolytic-uremic syndrome.30,31 Organ dysfunction is due to microvascular thrombosis. Commonly involved organs are the brain, kidneys, heart, and pancreas. It is critical to establish the diagnosis quickly because TTP can be rapidly fatal if not properly treated. Once the diagnosis of TTP is thought to be likely, therapy should be initiated quickly because of the proclivity of the disease to progress rapidly. All patients should initially be treated in the ICU. Platelet transfusions should be avoided except for life-threatening bleeding because of anecdotal reports of acute decompensation after platelet transfusions. A large-bore catheter will need to be placed, even in the face of severe thrombocytopenia, because the mainstay and only evidence-based component of therapy for TTP is plasma exchange.32,32a Most commonly, 1.5 plasma volumes are exchanged daily with FFP. A clue that TTP is the correct diagnosis is the color of the plasma removed from the first plasmapheresis. A red or brown color suggests free hemoglobin from intravascular hemolysis. If plasma exchange cannot be initiated in a timely manner, infusion of 30 mL/kg of FFP daily can be a temporizing maneuver.32 Corticosteroids (e.g., prednisone, 1 mg/kg/day) are commonly used as well.32a The use of antiplatelet agents, which may increase the risk of bleeding, and vincristine is controversial.33 In patients who do not respond to initial therapy, rituximab is an attractive second-line therapy.30,34 Hemolytic-uremic syndrome (HUS), the triad of thrombocytopenia, MAHA, and acute kidney injury, is most commonly due to infection with Shiga toxin–producing Escherichia coli in children.35 Adult atypical HUS (aHUS) is not Shiga toxin associated. It may be idiopathic or associated with calcineurin inhibitors (cyclosporin, tacrolimus), pregnancy, and HIV. aHUS is associated with low C3 and is a complement deposition disease. Mutations in complement factor H (CFH) are found in a minority of cases. Although not as efficacious as when used for TTP, plasma exchange is used for aHUS. The anti-C5 antibody eculizumab (Soliris) has been FDA approved for the treatment of aHUS.36 Plasma exchange should be initiated while waiting to obtain eculizumab. Supplemental eculizumab dosing is required after each plasma exchange.37 Patients need to receive quadravalent meningococcal vaccine before starting eculizumab.
Hemorrhagic and Thrombotic Disorders
Laboratory Tests of Coagulation
Disorders of Platelets
Thrombocytopenia
Mechanisms and General Management
Pseudothrombocytopenia
Drug-Induced Thrombocytopenia
Glycoprotein IIb/IIIa Inhibitors
Immune Thrombocytopenic Purpura
Post-transfusion Purpura
Acquired Platelet Dysfunction
Medication-Induced Abnormalities
Renal Failure
Complex Thrombohemorrhagic Disorders
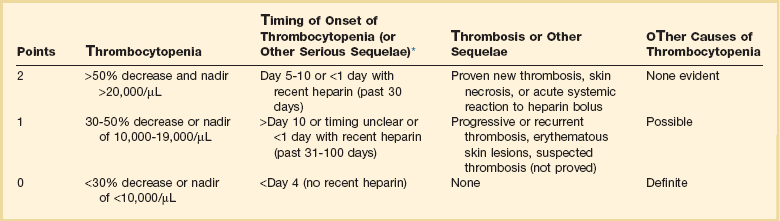
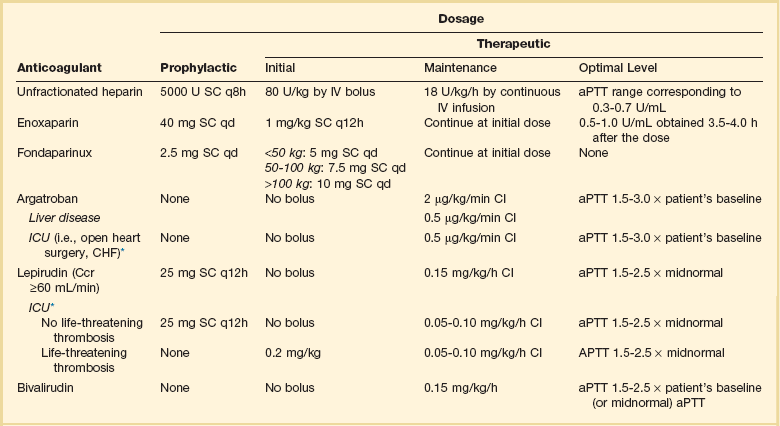
Heparin-Induced Thrombocytopenia
Thrombotic Thrombocytopenic Purpura
Hemolytic-Uremic Syndrome

Full access? Get Clinical Tree


Anesthesia Key
Fastest Anesthesia & Intensive Care & Emergency Medicine Insight Engine