Fig. 73.1
Chest radiograph on admission (a) demonstrated bilateral infiltrates, which corresponded to bilateral ground glass opacities and interstitial thickening on a CT performed the same day (b). After 1 month, pulmonary infiltrates were consistently more severe, despite multiple courses of antibiotics and diuresis (c). After treatment with etoposide and dexamethasone, infiltrates resolved by the time of discharge (d)
He presented to the hospital for the third time 2 days after discharge with hypotension and hypoxia requiring endotracheal intubation and vasopressors, and antibiotics and steroids were restarted. He was admitted to the ICU and his pulmonary status rapidly improved. After 3 days, however, he again decompensated with hypotension and an elevated lactate. He received intravenous fluids and intravenous steroids. Given the unclear association between changes in his antimicrobial regimen, steroid dosing, and clinical condition, his steroid dose was decreased from 40 mg prednisone daily by 10 mg each day. A repeat bone marrow biopsy was performed, which showed rare hemophagocytic macrophages but no evidence of malignancy. The next day, in the setting of reducing his steroid dose, he again had an episode of hypoxia and hypotension. Ferritin was measured at that time and was 9148 ng/mL, increased from 4074 ng/mL the day before (Fig. 73.2). sIL2-R was measured and was elevated at 3494 U/mL. A diagnosis of HLH was made, and he was started on chemotherapy with etoposide and high dose dexamethasone. Eight weeks had elapsed since his initial presentation.
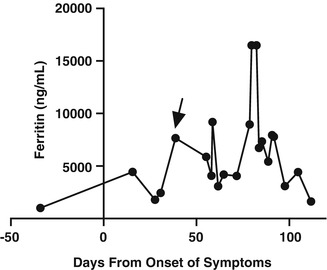
Fig. 73.2
Degree of hyperferritinemia varied significantly over the patient’s hospital course, with marked elevations at the time of diagnosis (arrow) and relapse of HLH
Question
How is hemophagocytic lymphohistiocytosis treated?
Answer
The patient was initiated on etoposide and dexamethasone without cyclosporine per the HLH-94 protocol. He rapidly improved and was moved to the hematology/oncology ward. His course was complicated by recurrent fevers and rising ferritin with the addition of filgrastim to his regimen after 3 weeks of therapy. This was discontinued and he improved uniformly from then on. After 1 month he was transferred to the physical medicine service for rehabilitation, and was subsequently discharged to home. He continued to do well 2 months after his diagnosis, and was preparing for allogeneic bone marrow transplant at that time.
HLH is a group of disorders characterized by aberrant immune activation leading to uncontrolled inflammation and, in many cases, critical illness. The underlying causes of HLH have been divided into primary causes, which encompass genetic defects in immune system regulation and often present in the neonatal period or early childhood, and secondary causes, which may be related to autoimmune disease, viral infection, or malignancy [1, 2]. However, there is growing appreciation that many adolescent and adult patients develop HLH due to the interaction of an underlying genetic defect with an environmental trigger, such as viral infection with Epstein-Barr virus or cytomegalovirus [3].
Presentation
In adults, HLH often presents with dramatic, but nonspecific, features of including fever, organomegaly, cytopenias, kidney injury, disseminated intravascular coagulation and altered mental status [4, 5]. As illustrated in the case above, in our experience rapid fluctuating hemodynamic instability and hypoxic respiratory failure are also common features resulting from the abnormal inflammatory response. These presenting symptoms and laboratory findings overlap significantly with severe sepsis. It is not uncommon for adult patients with HLH to have recently been hospitalized for presumed sepsis, and HLH is entertained as a diagnosis after one or more unexplained relapses. The most important step in recognizing HLH in the critically ill patient, therefore, is to consider it in the differential diagnosis alongside more common causes of shock and respiratory failure.
Diagnosis
While primary HLH is a genetic disorder and may be diagnosed by molecular testing with a suggestive family history and clinical presentation, secondary HLH is a clinical syndrome with diverse causes. Diagnostic criteria for HLH are currently based on the selection criteria for the HLH-2004 trial, which enrolled pediatric patients (Table 73.1, [6]).
Table 73.1
Diagnostic criteria for HLH derived from the HLH-2004 study
The diagnosis of HLH may be established by |
A. Molecular diagnosis consistent with HLH |
Or |
B. Five of the 8 criteria listed below: |
1. Fever >38.5 C |
2. Splenomegaly |
3. Cytopenias affecting 2 of 3 lineages in peripheral blood (RBC, platelets, or neutrophils) |
4. Hypertriglyceridemia or hypofibrinogenemia |
5. Hemophagocytosis in biopsy of the bone marrow, spleen, or liver |
6. Low or absent NK cell activity |
7. Hyperferritinemia |
8. Elevated sCD25 (soluble IL-2 receptor) |
The presence of fever, organomegaly, cytopenia, and hypertriglyceridemia or hypofibrinogenemia, while nonspecific, is widely accepted as helpful criteria in defining the syndrome. The presence of hemophagocytosis, despite lending HLH its name, is also nonspecific [7]. The search for appropriate biomarkers, then, remains an area of active and important investigation, as detailed below. In practice, the most critical information leading to the diagnosis of HLH is often derived from following the patient over several days, observing the response, or lack thereof, to treatment for sepsis or other common conditions, and the relationship of biomarkers to the patient’s evolving clinical condition.
Related posts:






Full access? Get Clinical Tree


