Hematologic/Oncologic
21-1 Pernicious Anemia
Robert Hendrickson
Clinical Presentation
Patients with pernicious anemia may present with symptoms of anemia (fatigue, lightheadedness, end-organ ischemia) or vitamin B12 deficiency (neuropathy, diarrhea). Physical examination may reveal macroglossia (large, red, smooth tongue), angular cheilitis, or “spoon nails.”
Pathophysiology
Pernicious anemia is a disorder in which vitamin B12 deficiency anemia results from atrophic gastritis and malabsorption. It is most common in women older than 60 years of age.1 Vitamin B12 is important in the growth and division of cells. Deficiency in vitamin B12 can lead to apoptosis of red blood cells (RBCs) and the release of immature, large reticulocytes and RBCs from the bone marrow (macrocytic anemia with numerous reticulocytes). Pernicious anemia develops secondary to destruction of gastric parietal cells due to autoantibodies and resulting malabsorption of vitamin B12.1
Diagnosis
The diagnosis should be considered if a macrocytic anemia is discovered in a patient who has the typical characteristics of pernicious anemia (e.g., older age, female, macroglossia). Patients with pernicious anemia have decreased vitamin B12 concentration, normal folate concentration, and a macrocytic anemia with thrombocytopenia.1 A Schilling test may be performed to determine whether the deficiency is the result of malabsorption.
Clinical Complications
Complications include the complications of anemia (myocardial infarction, cerebrovascular accident, syncope), as well as chronic diarrhea, peripheral neuropathy, and subacute combined degeneration of the spinal cord and cerebrum (potentially irreversible demyelination).
Management
Patients with pernicious anemia require intramuscular vitamin B12 supplementation monthly. Follow-up should be arranged for repeated evaluation.
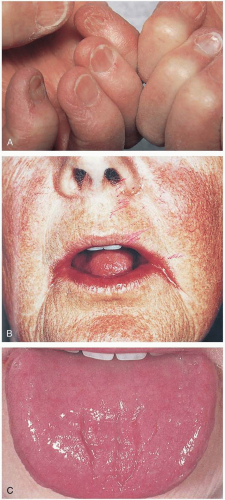 FIGURE 21-1 A: Koilonychia. These nail changes are characteristic of iron deficiency and consist of spooning concavity, longitudinal ridging, and brittleness. B: Angular cheilitis. Inflammation at the angles of the mouth is commonly associated with deficiencies of the B vitamins. C: A raw, fissured tongue, especially in the setting of peripheral neuropathy, should raise the suspicion of vitamin B12 deficiency. (A-C From Yamada, with permission.) |
REFERENCES
1. Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N Engl J Med 1997;337:1441-1448.
21-2 Sickle Cell Anemia
Robert Hendrickson
Clinical Presentation
The majority of patients with sickle cell anemia who present to the emergency department know that they have a diagnosis of sickle cell disease, because the clinical symptoms begin at a young age. Occasionally, patients with sickle cell variants (sickle β-thalassemia, sickle cell C disease) present in their teenage years. Patients usually are asymptomatic between sickling episodes. Stress, dehydration, trauma, hypoxia, or infection can trigger red blood cells (RBCs) to sickle and produce symptoms of end-organ ischemia. Symptoms of vaso-occlusive crisis can range from severe pain in muscles or bones to cerebrovascular accident (CVA) and pulmonary infarction (acute chest syndrome). The splenic sequestration syndrome may produce abdominal pain, anemia, and splenic rupture. Other symptoms are related to venous obstruction or ischemia and include priapism, avascular necrosis, hand-and-foot syndrome, renal papillary necrosis, and aplastic crisis (infarction of the bone marrow). Older children and adults are at risk of infection from encapsulated organisms.1
Pathophysiology
Sickle cell anemia is an autosomal codominant hereditary disorder of hemoglobin that produces malformation (“sickling”) of RBCs. A genetic substitution in the β-globin gene produces an altered hemoglobin, HbS. HbS polymerizes on deoxygenation, resulting in a rigid, sickle-shaped, dehydrated RBC with increased affinity for the endothelial wall.1 Sickling leads to vascular occlusion and end-organ ischemia.
Diagnosis
Most patients know their diagnosis and those of their family members; however, the diagnosis may be confirmed or clarified by hemoglobin electrophoresis.1
Clinical Complications
Complications include ischemia of any organ, CVA, myocardial infarction, splenic infarction, renal failure, acute chest syndrome, hemolysis, anemia, bony infarction, and sepsis from encapsulated organisms.1
Management
Patients with vaso-occlusive crises should be hydrated (orally or intravenously) and given adequate pain medication. Patients with pulmonary complaints or hypoxemia should receive supplemental oxygen and a chest radiograph to rule out acute chest syndrome. A complete blood count and reticulocyte count are helpful in ruling out aplastic crisis and determining the extent of anemia. For patients with fever, a search should be made for the source of infection, and cultures and antibiotic therapy should be started quickly.1 Splenic sequestration is an emergency that requires urgent exchange transfusion and possibly splenectomy.1 Patients with acute chest syndrome should be treated for a pulmonary infection and may require further workup and treatment for a pulmonary thrombosis. Admission should be considered for patients with central nervous system, pulmonary, or infectious crises; those who are unable to take oral fluids; and those with splenic sequestration or aplastic crisis.1
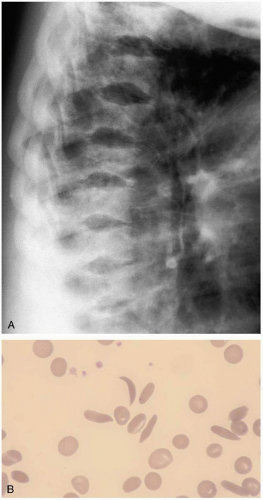 FIGURE 21-2 A: Fishmouth vertebrae, characteristic of long-standing sickle cell disease. (© David Effron, MD, 2004. Used with permission.) B: Drepanocyte (sickle cell). (From Anderson, with permission.) |
REFERENCES
1. Ballas SK. Sickle cell anaemia: progress in pathogenesis and treatment. Drugs 2002;62:1143-1172.
21-3 The Hemophilias
Robert Hendrickson
Clinical Presentation
Patients with hemophilia present with complaints that are related to bleeding. Traumatic or atraumatic hemarthrosis and muscle bleeds are a common phenomenon and can be exceedingly painful. Other bleeding complications include central nervous system (CNS) bleeds, gastrointestinal (GI) bleeds, and exsanguinating dermal or mucosal bleeding.1
Pathophysiology
The hemophilias are inherited disorders of coagulation caused by deficiencies of coagulation factors. Hemophilias A, B, and C are caused by deficiencies in factors 8, 9, and 11, respectively. Hemophilia C is typically a mild disorder with minimal risk of serious bleeding.1 Hemophilias A and B result from abnormalities in the long arm of the X chromosome. Circulating factor concentrations vary and are indirectly proportional to the severity and frequency of complications. Activated factors 8 and 9 are necessary components in the coagulation cascade and are required for amplification of activated factor 10.1 Deficiencies in these factors prevent the stabilization of platelet clots and may lead to bleeding.1
Diagnosis
Severe hemophilia A or B usually manifests early in life (before 4 years of age), whereas milder forms may be diagnosed later in life. Two thirds of patients have a family history.1 Patients with suspected hemophilia should be referred to a hematologist or primary doctor for definitive diagnosis.
Clinical Complications
Complications include exsanguinating mucosal or dermal hemorrhage, CNS hemorrhage, and GI hemorrhage. Patients with recurrent hemarthrosis and muscle bleeds may develop debilitating arthropathy and muscle atrophy.1 In the early 1980s, only pooled plasma-derived factor replacement was available, and thousands of hemophiliacs developed human immunodeficiency virus (HIV) infection, hepatitis A, and hepatitis B from the blood supply. With the current use of recombinant factors, there is no risk of disease transmission. However, plasma-derived factors are still used, and, although there is no risk of HIV, hepatitis B, or hepatitis C transmission, there remains a risk of parvovirus and hepatitis A transmission.1
Management
Bleeding in a patient with hemophilia is an emergency and should be treated promptly. Patients with mild hemophilia A (or von Willebrand’s disease) who present with mild bleeding (e.g., hemarthrosis) may be given desmopressin to raise the factor 8 concentration. More serious bleeding or severe underlying hemophilia may be treated with recombinant factor replacement.1
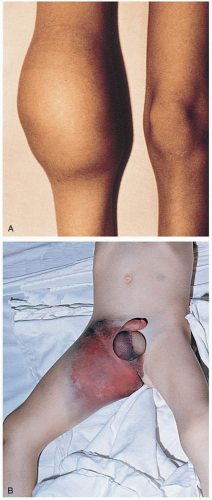 FIGURE 21-3 A: Hemophilic arthropathy. This figure illustrates the sequelae of recurrent joint bleeding. B: Large dissecting hematoma of the thigh in a patient with hemophilia A. The lesion resulted from a slight bump to the inguinal area and spread to involve the entire thigh. (A and B, from Greer, with permission.) |
REFERENCES
1. Bolton-Maggs PH, Pasi KJ. Haemophilias A and B. Lancet 2003;361:1801-1809.
21-4 Lymphoma
Robert Hendrickson
Clinical Presentation
Patients with lymphoma may present with painless peripheral lymphadenopathy (most common), splenomegaly or abdominal mass, weight loss, night sweats, fatigue, anemia, fever, or pruritus.1
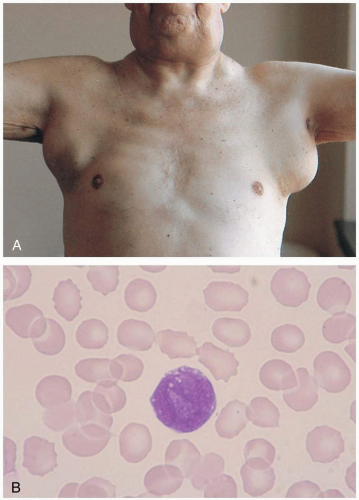 FIGURE 21-4 A: Malignant adenopathy of lymphoma. The nodes are larger than 2 cm in diameter, multiple, rubbery, and nontender. (From Handin, with permission.) B: Large follicular cell of B-cell origin (non-Hodgkin’s lymphoma). (From Anderson, with permission.)
Related posts:
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|





