Anemia
Anemia, or erythropenia, is the most common hematologic abnormality diagnosed and managed in the
PICU. The most

common anemia etiologies that require emergent evaluation

and care are acute blood loss (hemorrhage), acute hemolysis, and acute splenic sequestration. With acute blood loss (whether traumatic, surgical, or gastrointestinal), the approach is uniform: identify the potential source of bleeding, develop a diagnostic and/or therapeutic plan, and maintain intravascular volume with red blood cell (
RBC) transfusion and crystalloid until the plan can be implemented. In addition, chronic inflammation and frequent phlebotomy contribute to more insidious acute anemia that frequently requires transfusion
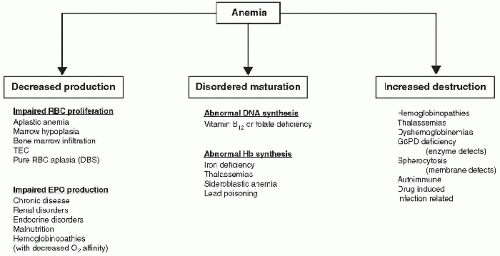
and may not be avoidable. However, when an acutely ill child presents with anemia among other symptoms and problems, the critical care provider must work through the primary and secondary causes for the anemia, which involves evaluating the potential etiologies by considering the different mechanisms: (a) conditions of failed
RBC production, (b) conditions of increased
RBC destruction, and (c) conditions of abnormal
RBC maturation (
Fig. 116.1).
Anemia, regardless of etiology, results in a decreased O
2-carrying capacity and, when severe, can result in decreased O
2 delivery. Physical symptoms are extremely variable and not predictive of anemia severity (
1). Common symptoms of
anemia are pallor, nausea, vomiting, weakness, fatigue, irritability, tachycardia, tachypnea, and edema. Although anemia is a common reason for
RBC transfusion, in the
PICU other factors are often involved (
2). In addition, comorbid conditions that may contribute directly to the anemic state are both acute and chronic in nature. Chronic illnesses with anemia as a feature often have adaptive pathophysiology that alters vascular perfusion and diphosphoglycerate levels, expands plasma volume, and lowers blood viscosity.
Anemia secondary to acute hemorrhage and sickle cell disease (
SCD) are discussed in
Chapters 28 and
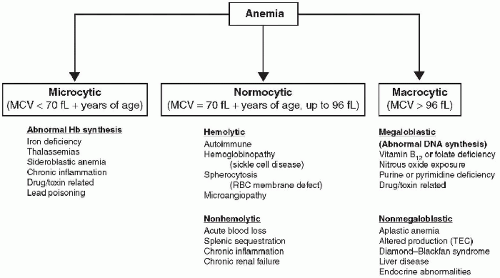
119. The differential diagnoses of anemia based on mean corpuscular volume (MCV) are summarized in
Figure 116.2. Following are some of the most commonly encountered causes for reduction in
RBC number and function in children.
Iron-deficiency anemia is the most common nutritional abnormality worldwide and is the leading cause of anemia in early childhood. When iron-deficiency anemia is severe (e.g., hemoglobin [
Hb] is < 4 g/dL), children can be symptomatic with cardiac and respiratory instability. Diagnosis of iron-deficiency anemia typically has low
Hb, low
RBC count, low MCV, and low reticulocyte count, with elevated free erythrocyte protoporphyrin and red-cell distribution width. The ferritin is depressed and serum iron is low with an elevated total-iron-binding capacity and transferrin. The peripheral blood smear demonstrates microcytic and hypochromic erythrocytes. During the first 4-6 months of life, most full-term infants are iron sufficient owing to transplacental passage of iron during the last trimester of pregnancy. Iron deficiency occurs most frequently in children <3 years of age and results from inadequate iron intake during this period of rapid growth. Poor iron intake is often complicated by other clinical findings of poor nutrition, neglect, and/or excessive dietary intake of cow’s milk. In older children and adults with iron deficiency, occult blood loss should be suspected and should prompt a gastrointestinal evaluation. Differentiating between the two most common causes of microcytic anemia (iron deficiency vs. thalassemia) can be challenging. A low ferritin is diagnostic of iron deficiency, while an elevated HbA
2 is specific for beta thalassemia trait. However, the ferritin is
an acute-phase reactant and may be elevated with inflammation. The Mentzer index (MCV/
RBC count) can be a useful bedside tool. When applied to conditions of microcytic anemia, Mentzer index values of >13.5 suggest iron deficiency, while a value of <11.5 suggests thalassemia minor. Differential diagnosis for microcytic anemia also includes lead poisoning, chronic inflammation, copper deficiency, and thalassemia (
Fig. 116.2).
In addition to low ferritin, perhaps the most conclusive evidence of iron-deficiency anemia is increasing
Hb during therapeutic iron supplementation. Optimal response for
treatment is obtained with 3-6 mg/kg/d of elemental iron. Enteral ferrous sulfate is the preferred form; parenteral administration (with iron dextran) is an option if compliance or tolerance of the oral preparation is an issue. Recommendations for
prevention of iron-deficiency anemia include iron supplementation (1-2 mg/kg/d) for all breastfed infants after 4 months of age, use of iron-fortified formulas (containing 12 mg iron as ferrous sulfate per liter) and cereals, iron supplement (2-3 mg/kg/d) to preterm infants after the first month of life, and delaying the introduction of cow’s milk until after 1 year of age.
Blood transfusion is rarely needed in the treatment of iron-deficiency anemia (
3). However, studies of children with multifactorial anemia (nutritional, malaria, hemoglobinopathy, etc.) in Africa have demonstrated significant reduction in mortality with blood transfusion therapy, regardless of age. These data, along with published clinical practice, supports transfusing children with or without cardiorespiratory symptoms when
Hb is ≤4 g/dL (
4). In infants and children who have
Hb values between 4 and 6 g/dL, treatment practices vary worldwide and the decision to transfuse should be based on symptoms, available resources, comorbid conditions (malaria, fever), and end-organ manifestations. If transfusion therapy is undertaken in either severe or moderate anemia, slow, monitored transfusion of packed RBCs should be performed, with continuous cardiorespiratory monitoring to avoid congestive heart failure from the adaptive changes of prolonged anemia (
3). Typically, packed RBCs (5 mL/kg) should be administered over 4 hours, followed by another 5 mL/kg transfusion over 4 hours. Diuretics may also be required to avoid volume overload in an anemic patient’s volume-expanded state.
Hemolytic anemia and autoimmune hemolytic anemia can be caused by either intracellular or extracellular disorders. Intracellular disorders consist of
RBC membrane defects, abnormal erythrocyte metabolism, or hemoglobinopathies. Extracorpuscular disorders include immune-mediated destruction, mechanical fragmentation, infections, drugs, chemicals, and venoms. Inherent red-cell susceptibilities, such as enzyme disorders and unstable hereditary hemoglobinopathies, enhance the ability of certain agents to produce damage. For instance, the hexose-monophosphate shunt and the glutathione pathways are the predominant mechanisms by which the erythrocyte handles oxidative stress. Disorders of either of these pathways can result in oxidative damage to the
RBC and lysis (
5). Discussion will focus on the most common intracellular disorder (glucose-6-phosphate dehydrogenase [
G6PD] deficiency), and the most common extracellular disorder (autoimmune hemolytic anemia). Hemolytic uremic syndrome is another important cause of hemolytic anemia in the
PICU. Tests that are useful in the diagnosis of a hemolytic process include evaluation of peripheral blood smear for irregular RBCs (spherocytes or schistocytes), and additional investigation for Heinz bodies (denatured
Hb), elevated reticulocyte count, positive Coombs test (direct and indirect), elevated serum aspartate aminotransferase, lactate dehydrogenase, and serum bilirubin, with lowered serum haptoglobin.
G6PD deficiency is the most common RBC-associated enzyme disorder and is genetically determined by X-linked inheritance. Children and adults prone to this disorder include Africans, Asians, and those of Mediterranean descent. The Mediterranean variety is the classic form and is the most severe, as it results in the lowest
G6PD activity. The African variety is unique because only the mature RBCs are enzymedeficient. The incidence of
G6PD deficiency in African American males is ˜11% (
6). The susceptibility of a patient with
G6PD deficiency to drug-induced oxidative stress depends on the phenotype of the enzyme disorder and on drug metabolism and excretion.
G6PD catalyzes the reduction of nicotinamide adenine dinucleotide phosphate (
NADP) to NADPH in the hexose-monophosphate shunt. NADPH converts glutathione disulfide to reduced glutathione. Glutathione, in turn, inactivates hydrogen peroxide (H
2O
2) and protects protein sulfhydryl groups from oxidation. Lack of this enzyme allows for the rapid depletion of protective antioxidants and subsequent denaturation of the
Hb unit. Oxidative stressors attack
Hb sulfhydryl groups, releasing heme, the protein chain of which then unfolds and precipitates as insoluble aggregates (Heinz bodies). Unripened peaches, fava beans, methylene blue, naphthalene, phenazopyridine, nitroglycerin, prilocaine, benzocaine, and sulfamethoxazole are just a few of the agents that result in hemolysis in G6PD-deficient patients. Salicylates do not pose a risk for hemolysis, except in high doses. Avoidance of oxidants (e.g., sulfa drugs) and careful observation during stress (e.g., infection and surgery) are necessary. Patients with a diagnosis of
G6PD deficiency who develop severe pallor or abdominal pain require immediate evaluation. Other patients who develop hemolytic symptoms of unknown etiology should have a
G6PD assay performed. Identification of possible sources that trigger hemolysis and the elimination of continued exposure are crucial to the patient’s recovery.
RBC transfusion is sometimes necessary during episodes of severe hemolysis.
Antibody-mediated (autoimmune) hemolytic anemia is an extracellular process of
RBC destruction. Typically, this disorder manifests as sudden pallor and fatigue, often following a viral illness. Because of the rapid onset, jaundice, hyperbilirubinemia, and reticulocytosis might not be present initially. The laboratory findings reveal a rapidly falling
Hb, low haptoglobin concentration, increased bilirubin metabolites in the urine, positive Coombs tests, and abundant spherocytes on the smear. The direct Coombs test confirms the diagnosis of antibody bound to the patient’s RBCs. The indirect Coombs test detects free antibody in the patient’s serum. Clinically significant hemolysis can occur with either
IgG or
IgM antibodies, but most commonly with
IgG. Typically,
IgG antibodies do not agglutinate
RBC in vitro (at 22°C) and bind RBCs maximally at 37°C; therefore, they are termed
warm antibodies. In contrast,
IgM antibodies cause in vitro agglutination at ≤20°C and are termed
cold antibodies. Because
IgM antibodies do not bind red cells avidly at physiologic temperatures, they usually do not cause clinically significant hemolysis.
IgG antibody-coated RBCs are destroyed in the reticuloendothelial system of the spleen rather than intravascularly. The treatment involves hospitalization, careful observation, high-dose steroids, and, if necessary,
RBC transfusion using the most compatible blood. Additional therapies such as plasmapheresis and/or splenectomy are options for refractory patients.
Transient erythrocytopenia of childhood (TEC) is characterized by the gradual onset of a normocytic anemia and reticulocytopenia caused by temporary suppression of
RBC production. It usually occurs in children between 6 months and 3 years of age and is commonly preceded by a viral illness. Full recovery usually occurs in 4-6 weeks. Blood transfusions may be required if the
Hb concentration is <4 g/dL and/or the patient develops symptomatic anemia. Follow-up studies are necessary until the
Hb concentration and reticulocyte count recover. In ˜25% of patients, the anemia is associated with mild neutropenia.
TEC must be distinguished from a rare disease, termed
pure red-cell aplasia or
Diamond-Blackfan syndrome. Patients with Diamond-Blackfan syndrome typically have macrocytic erythrocytes (elevated MCV), dysmorphic features, and persistent anemia. Bone marrow recovery is the hallmark of
TEC and rules out Diamond-Blackfan syndrome.
Bone marrow infiltration can result from malignant cells or be the result of other processes such as inherited metabolic disorders or infections (fungus, tuberculosis). Regardless of the primary process, the result is usually a normocytic anemia (normal MCV) with a low reticulocyte count. These patients can have lymphadenopathy, hepatomegaly, and splenomegaly on physical examination, although laboratory abnormalities can include reticulocytopenia, neutropenia, thrombocytopenia, and circulating immature cells (e.g., nucleated RBCs, promyelocytes, metamyelocytes, and myelocytes). Bone marrow examination is essential in this setting to establish the correct diagnosis.
Acquired aplastic anemia is defined as peripheral blood pancytopenia with bone marrow hypocellularity in the absence of underlying malignant or myeloproliferative disease (
7,
8). The principal pathophysiologic mechanism is thought to be immune-mediated destruction of hematopoietic stem cells by cytotoxic T lymphocytes. The incidence of acquired aplastic anemia increases with age. The incidence is low for patients <1 year of age, slowly increases to an intermediate level by the age of 50 years, and has its highest occurrence in older adults (>50 years of age). Drug-induced causes account for 50% of all cases (
7,
8).
Drug-induced aplastic anemia can result from either toxic effects or immune-mediated phenomena. Progenitor stem cells are most commonly affected. Common toxic agents include ionizing radiation, chemotherapeutic drugs, antibiotics (chloramphenicol), hydrocarbons (benzene), anti-inflammatory medications (phenylbutazone, indomethacin), and metals (gold) (
Table 116.5). Other drugs with a relatively high risk for aplastic anemia are anticonvulsants (fosphenytoin, phenytoin, carbamazepine) and quinacrine (
9). Phenytoin and carbamazepine result in the production of toxic areneoxide intermediate metabolites in vivo that bind covalently to macromolecules of marrow stem cells and lymphocytes. Supportive care is typically required, with recovery of the
RBC counts occurring over time once the causative agent is discontinued.
Megaloblastic anemia may be caused by decreased vitamin absorption, impaired metabolism, or both. Vitamin B
12 and folate deficiencies selectively impair cellular
DNA synthesis. Because RNA protein production is maintained, an increased ratio of cytoplasmic-to-nucleic mass is created resulting in megaloblastic RBCs (including granulocytes and megakaryocytes) (
10). Abnormal cellular maturation leads to premature cellular death. Decreased vitamin B
12 absorption is rare in children and usually results from limited nutritional intake or intestinal malabsorptive diseases. Agents that impair enteral absorption of vitamin B
12 include metformin, colchicine, neomycin, para-aminosalicylic acid, and slow-release potassium chloride. Causes of folate deficiency include sepsis, pregnancy, malignancy, chronic hemodialysis, and medications.
Some anticonvulsant medication can lower serum folate concentrations by limiting enteral absorption. Approximately 50% of patients on long-term phenytoin therapy have low serum folate concentrations and ˜30% have red-cell macrocytosis and early megaloblastic changes in the bone marrow. Folate replacement therapy improves the anemia. Methotrexate, trimethoprim, triamterene, and pyrimethamine inhibit dihydrofolate reductase, and megaloblastic anemia, which is not folate responsive, can develop during long-term and high-dose therapy. Acute megaloblastic anemia may also be caused by prolonged or repeated nitrous oxide anesthesia (
11). This gas inactivates vitamin B
12 through oxidation. The effect on
RBC formation is usually observed after a cumulative 5-hour exposure to ≥50% nitrous oxide. Case reports have also suggested that nitrous oxide exposure contributed to acute megaloblastic anemia and neuropathy in susceptible patients with comorbid illnesses such as viral infections, poor nutrition, alcoholism, malignancy, and sepsis (
6).
Hemoglobinopathies
Primary hemoglobinopathy is a term that describes structural abnormalities of
Hb that are often inherited. The globin genes are found in clusters on chromosomes 11 (
β-globin chain) and 16 (
α-globin chain). Point mutations and deletions in the genes for the
Hb molecule result in decreased expression or production of functionally abnormal
Hb. In normal human maturation, HbA (
α2
β2) becomes the dominant form, taking over from the
HbF (
α2
ρ2) during the first year of life. The thalassemias are the most common worldwide genetic disorder. Examples of common beta-chain mutations that result in hemoglobinopathies include S (sickle) and C, which can result in symptomatic anemia syndromes. Approximately 700 structural variants of
Hb have been described with the majority of these being extremely rare and asymptomatic. More unstable
Hb forms result in denaturation and formation of Heinz bodies on peripheral smear in the
RBC. While most mutations of
Hb are asymptomatic, some will present with mild, chronic anemia and others with increased levels of chronic and episodic hemolysis. Detection on routine
Hb electrophoresis is difficult and specialized testing is often required to detect these abnormalities.
Other structurally abnormal Hbs result in increased O
2 
affinity and relative tissue hypoxia, which leads to polycythemia. Erythropoietin levels are often elevated and the diagnosis is difficult.
Hb abnormalities that result in decreased O
2 binding result in cyanosis. A complete review is beyond the scope of this chapter; however,
familial methemoglobinemia (HbM) deserves comment. HbM is a rare autosomal-dominant disorder and affected individuals often present with cyanosis at birth. The structural abnormality results in stabilization of the iron heme moiety in the ferric (Fe
3+) form, which does not bind oxygen. The “secondary” causes of methemoglobinemia (
MetHb) are more common. In the secondary causes the oxidized form of
Hb can be converted back to the reduced form by enzymes in the blood (see below). Unlike the secondary causes, the diagnosis of HbM was classically made when individuals failed to respond to reduction by methylene blue therapy. HbM and the other clinically significant hemoglobinopathies are detectable by newborn screening utilizing high-performance
liquid chromatography or electrophoresis (as in
HbS). Chronic complications from HbM are rare. However, if hemolysis is chronic or acute, it can cause jaundice, cardiomegaly, hepatosplenomegaly, neurologic impairment, nephropathy, retinopathy, impaired growth and development, gallbladder stones, and skin ulcers. Diagnostic evaluation should include
CBC and reticulocyte count, bilirubin, pulse oximetry, blood pressure, growth and development, academic performance, biliary ultrasonography, and renal and pulmonary function studies.
Secondary hemoglobinopathies result from conditions that either induce abnormal
Hb production or adversely affect
Hb function. Dyshemoglobinemias are conditions that produce abnormal O
2 binding of structurally normal
Hb. Disorders of heme moiety oxidation occur in both genetically susceptible and unsusceptible individuals. Environmental and iatrogenic causes of
MetHb, carboxyhemoglobinemia, cyanohemoglobinemia, and sulfhemoglobinemia have been reported. Both chronic and acute forms have been described, with anemia, decreased O
2 delivery, and functional hypoxia as commonly associated features. Toxic exposures, such as cyanide and carbon monoxide, resulting in abnormal
Hb function are discussed in
Chapter 35.
Only gold members can continue reading.
Log In or
Register to continue
Related

Full access? Get Clinical Tree


 Hemorrhage, iatrogenic blood loss, and chronic inflammatory diseases are the most common causes of anemia; acute management of chronic causes of anemia is also often carried out in the PICU.
Hemorrhage, iatrogenic blood loss, and chronic inflammatory diseases are the most common causes of anemia; acute management of chronic causes of anemia is also often carried out in the PICU. Acute hemolysis and splenic sequestration are other causes of anemia that often require PICU admission and management.
Acute hemolysis and splenic sequestration are other causes of anemia that often require PICU admission and management. Altered O2-carrying capacity of acquired and congenital hemoglobinopathies impacts tissue oxygen delivery.
Altered O2-carrying capacity of acquired and congenital hemoglobinopathies impacts tissue oxygen delivery. Nonmalignant causes of abnormal (low and high) white blood cell (WBC) counts are common in the PICU.
Nonmalignant causes of abnormal (low and high) white blood cell (WBC) counts are common in the PICU.
 Thrombocytosis does not routinely require an intervention.
Thrombocytosis does not routinely require an intervention. The development of thrombocytopenia has been associated with increased morbidity, mortality, and length of stay in critically ill adults and children.
The development of thrombocytopenia has been associated with increased morbidity, mortality, and length of stay in critically ill adults and children. Immune-mediated thrombocytopenia in children and infants requires prompt recognition and treatment to reduce bleeding risk.
Immune-mediated thrombocytopenia in children and infants requires prompt recognition and treatment to reduce bleeding risk. Commonly used PICU medication can impact platelet function and number.
Commonly used PICU medication can impact platelet function and number. Thrombotic disease is less common in children than adults, but has been described in the pediatric population with increasing frequency and may be associated with significant morbidity and mortality.
Thrombotic disease is less common in children than adults, but has been described in the pediatric population with increasing frequency and may be associated with significant morbidity and mortality. Multiple risk factors for thrombosis are associated with critical care (vascular catheters, parenteral nutrition, heparin administration, etc.), and predisposing disease processes (cancer, nephrotic syndrome, and congenital heart disease).
Multiple risk factors for thrombosis are associated with critical care (vascular catheters, parenteral nutrition, heparin administration, etc.), and predisposing disease processes (cancer, nephrotic syndrome, and congenital heart disease). PICU physicians should be familiar with the evolving recommendations for the detection and treatment of thromboembolism in children.
PICU physicians should be familiar with the evolving recommendations for the detection and treatment of thromboembolism in children.