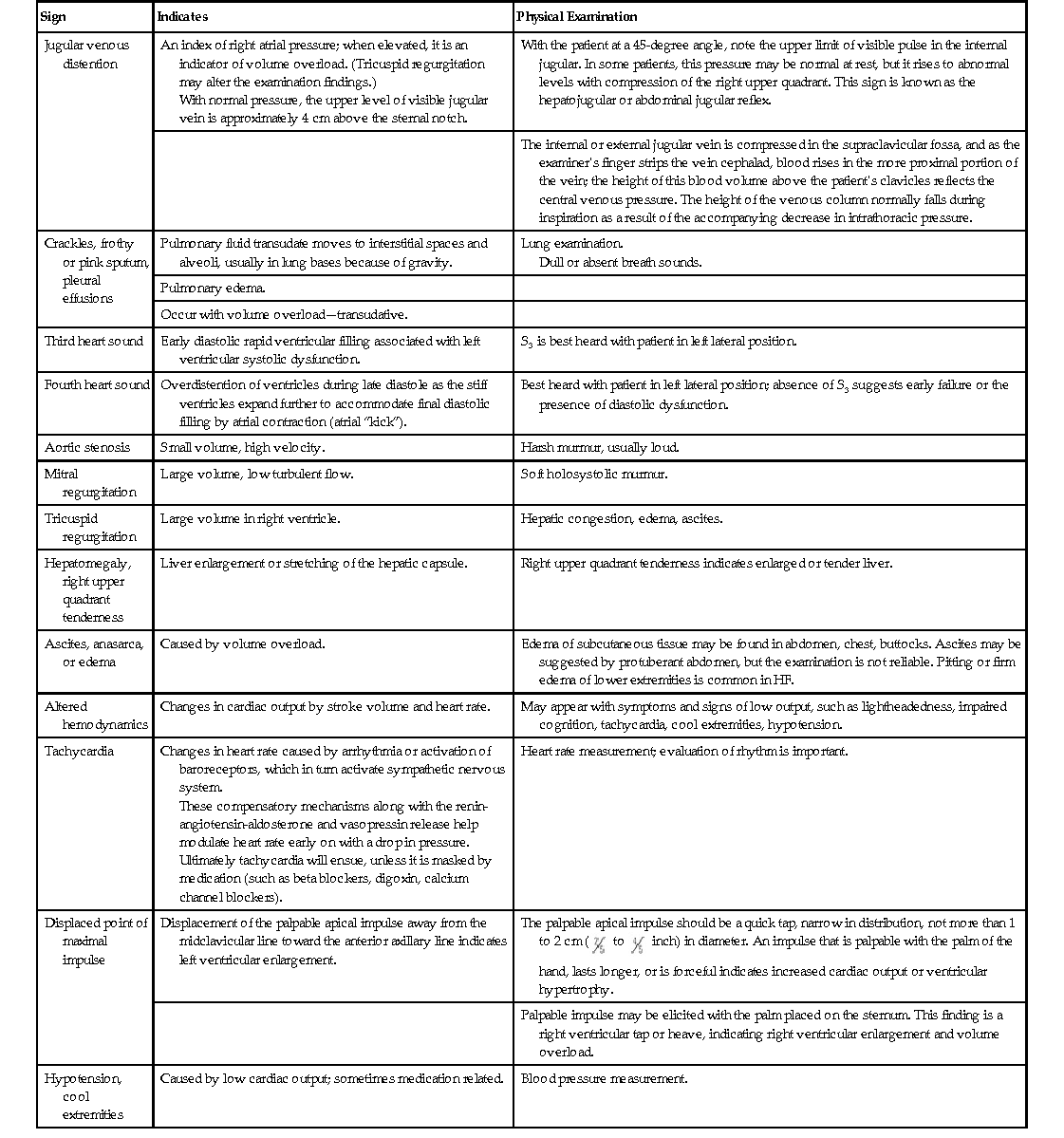
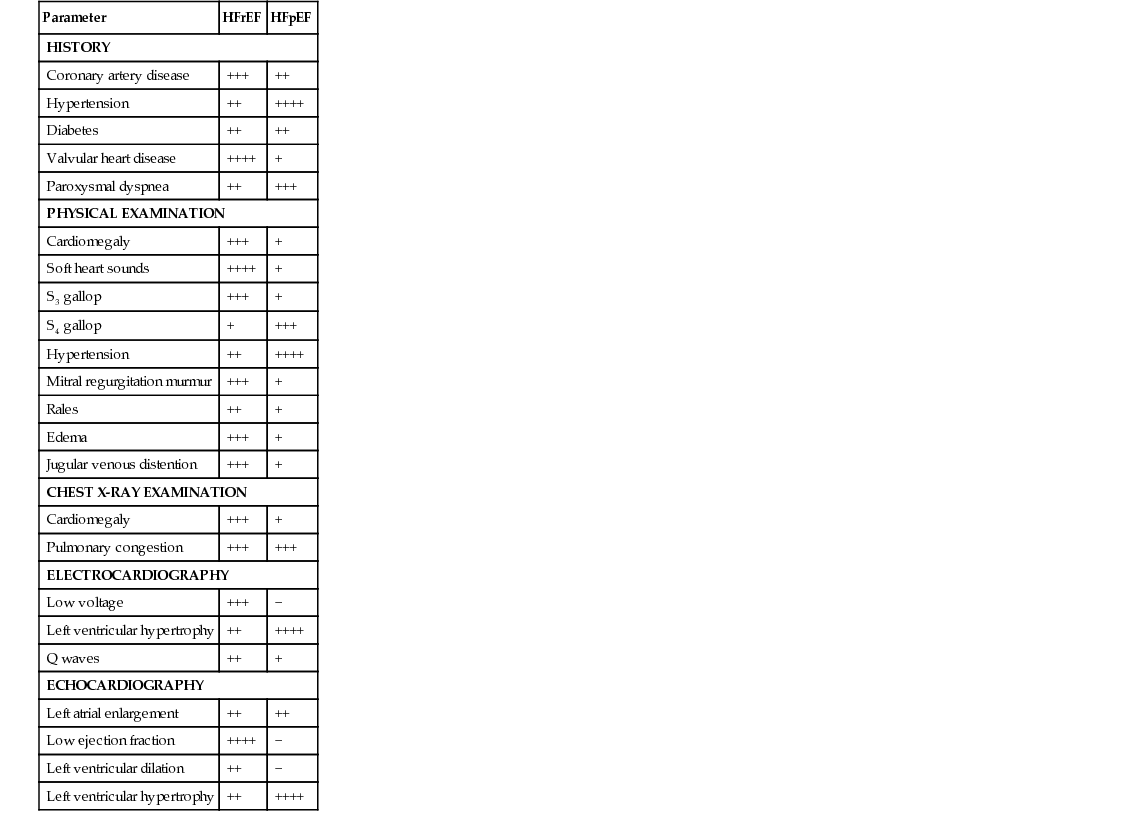
Virginia L. Beggs Heart failure (HF) is a complex syndrome characterized by the inability of the heart to meet the body’s metabolic demands; it is a clinical diagnosis. It results from any structural or functional cardiac disorder that impairs the ventricle’s ability to fill or to eject blood properly. HF is not the same as cardiomyopathy or left ventricular dysfunction (Table 121-1). HF has been divided into two main types: 2. HFpEF: heart failure with preserved ejection fraction of 50% or higher, associated with impairment of ventricular filling and relaxation, wherein the left ventricular filling pressures are often high, resulting in a reduced stroke volume with exertion, causing HF symptoms. HFpEF now accounts for approximately 50% of all patients with HF.1 TABLE 121-1 HFrEF Versus HFpEF in Heart Failure: Differences in History, Physical Examination Findings, and Diagnostic Test Results* Although coronary artery disease is the most common cause of HFrEF, hypertension, atrial fibrillation, and diabetes are common antecedents of HFpEF. It has been hypothesized that HFpEF may be more than just one problem. The Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF) registry shows a similar postdischarge mortality risk and rehospitalization rate for both HFpEF and HFrEF patients. Clinical presentation of HF is wide ranging, from mild, exertionally related dyspnea resulting from fluid retention to cardiogenic shock and lethal arrhythmias. Cardinal symptoms of HF are dyspnea and fatigue, often but not always associated with volume overload.2–4 The etiology of HF can be divided into the following three broad categories: The most common diseases associated with HF are coronary artery disease, hypertension, and dilated cardiomyopathies (Box 121-1). Most forms of heart disease predispose the patient to HF over time; viral, metabolic, and toxic insults to the myocardium can cause acute symptomatic HF that may become chronic if left untreated. Common antecedents of HFpEF include hypertension, diabetes, and atrial fibrillation. Early diagnosis and treatment can improve quality of life and life expectancy for people who have HF. The 30-day, 1-year, and 5-year case fatality rates after a hospitalization for HF are 10.4%, 22%, and 42.3%, respectively. The OPTIMIZE-HF registry shows a similar postdischarge mortality risk and rehospitalization rate for both HFpEF and HFrEF.4 Cardiomyopathy, the most common cause of HF, is a disease process of the myocardium that affects the heart’s pumping ability. Classification of heart muscle disease is complex; an expert panel defined cardiomyopathies as “a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation and are due to a variety of causes that frequently are genetic. Cardiomyopathies either are confined to the heart or are part of generalized systemic disorders, often leading to cardiovascular death or progressive heart failure–related disability.”5 Cardiomyopathies have been divided into two major groups: 1. Primary (including genetic, nongenetic, and acquired cardiomyopathies)—confined to the heart muscle 2. Secondary—heart muscle involvement as part of a general or systemic disease or disorder An example of a primary cardiomyopathy would be the genetic disorder hypertrophic cardiomyopathy; an example of an acquired cardiomyopathy would be myocarditis or an inflammatory cardiomyopathy. Stress cardiomyopathy (Takotsubo), infiltrative cardiomyopathy such as amyloidosis, and peripartum cardiomyopathy are other examples of secondary cardiomyopathy.5 HF is a societal epidemic in our country because of its incidence, prevalence, and high cost. HF is a leading cause of morbidity and mortality; 670,000 new cases are identified each year. The lifetime risk of HF for both men and women at 40 years of age is 1 in 5. Worldwide, 1% to 2% of the population has HF in developed countries; the prevalence approaches 10% in those older than 70 years. In the United States, the estimated direct and indirect cost of HF by 2030 is estimated to be $70 billion. This total includes the cost of health care services, medications, and lost productivity.4 Many individuals with HF have antecedent hypertension or myocardial infarction (MI). Other risk factors include coronary artery disease, diabetes, renal disease, obesity, smoking, and increasing age. African Americans have a higher prevalence of HF than other ethnicities and a greater 5-year mortality than whites. Hypertension is believed to account for some of the risk difference; socioeconomic factors that affect access to health care may be another important factor after control for conventional risk factors.4 HF is a clinical syndrome characterized by signs and symptoms of volume excess. Whereas there can be several causes of the HF, common pathophysiologic mechanisms characterize this important condition. Understanding these mechanisms is critical to selection of successful therapeutic interventions. Systolic dysfunction, which accounts for about half of all HF cases, is a decrease in both ejection fraction (40% or less) and cardiac output. In systolic dysfunction, when looking at a hemodynamic model, the three determinants of ventricular function—preload, contractility, and afterload—are usually altered. Preload is the degree of myocardial fiber stretch at the end of ventricular filling. When the heart ejects subnormally, there is an increased volume of blood left in the ventricular chambers (increased left ventricular end-systolic volume). This excess volume leads to distention of the ventricles and increased interventricular pressure at the onset of diastole. Filling must then occur at higher pressures during diastole. At small increases of volume and pressure, nonfailing myocardial fibers have the intrinsic property of increasing their force of contraction in an attempt to “revert” the subsequent volume and pressure conditions of both heart ejection and filling back to normal. This intrinsic property also enables the heart to maintain cardiac output during states of pressure or volume overload. However, in the failing heart, the failing myocardial fibers are both excessively overloaded and stretched beyond lengths commensurate with the normal reflex-increased force of contraction. This results in left ventricular remodeling, with dilation and impaired contractility, and activation of the sympathetic and renin-angiotensin-aldosterone systems. The ventricular dysfunction in HFrEF is accompanied by a decrease in myocardial contractility, a reduction in ejection fraction, and often a reduction in stroke volume and cardiac output. If HF is left untreated, symptoms develop and worsen, with declining functional capacity, recurrent acute decompensated events, life-threatening arrhythmias, and pump failure. Effective treatment of HF, primarily pharmacotherapy, depends on the interruption of left ventricular remodeling and the pathophysiologic processes that accompany it. Afterload is the amount of left ventricular wall tension that develops during systole; it is determined by both the size of the ventricular chamber and the dynamic vascular resistance against which the heart contracts. According to Laplace’s law, an increase in the radius of the ventricle results in an increase in wall tension. Because systolic blood pressure closely approximates afterload, it is a clinically important indicator of myocardial load or afterload. The LVEF is a function of afterload and an afterload-dependent measure of contractility. Chronic elevation of cardiac afterload can lead to ventricular enlargement, reduction in ejection fraction, and reduction in stroke volume and cardiac output. Several systemic mechanisms exist for the body to compensate for the reduction in cardiac output. Early on, these compensatory mechanisms serve to increase cardiac output and tissue perfusion. In the long run, however, they lead to further cardiac injury and further decompensation.1 The prevalence of HFpEF has increased dramatically. Controversy surrounds the true cause(s) of HFpEF. One mechanism believed responsible is increased ventricular stiffness and reduced compliance of the left ventricle, which produces a rise in cardiac filling pressures during diastole. Left ventricular distensibility is reduced during part or all of diastole, and filling pressures must increase to maintain a constant ventricular volume. Whereas filling pressures in the left ventricle are increased during both rest and exercise, the failure of a normal rise in cardiac output during exertion results in characteristic symptoms of HF, particularly dyspnea. The heart attempts to initially compensate for this impaired distensibility through the “booster” effect of augmented left atrial contraction, resulting over time in left atrial dilation. The incidence of HFpEF increases with age and is more prevalent in older adult women. Recent studies have examined the role of inflammation contributing to diastolic abnormalities. The most common factors associated with HFpEF are hypertension, ischemia resulting from coronary artery disease, aortic stenosis, and infiltrative or restrictive myocardial diseases.2 Several interrelated compensatory mechanisms attempt to maintain normal ventricular contractility, ventricular pressures, cardiac output, and blood pressure. The three primary compensatory mechanisms are increased sympathetic adrenergic activity with a resultant increase in circulating neurohormones, neuroendocrine activation of the renin-angiotensin-aldosterone system, and ventricular remodeling. In addition, as HF progresses, neurohormonal alterations in peripheral vasculature and renal function occur. Sodium and water retention through the renal tubules results in decreased renal perfusion and rising blood urea nitrogen and creatinine concentrations, broadly called cardiorenal syndrome, which can be chronic or acute. The same compensatory mechanisms in early and acute stages gradually fail as HF progresses, and they are responsible for the eventual deterioration in cardiac function.1 Baroreceptors and chemoreceptors in the heart and vascular system, sensitive to stretch, pH and CO2, help regulate blood pressure. Cardiac reflexes regulate heart rate. Abnormalities in both baroreceptor and cardiac reflexes have been documented in HF.1 In a healthy heart, stimulation of the baroreceptor reflex results in activation of the parasympathetic nervous system and inhibition of the sympathetic nervous system. Heart rate and systemic vascular resistance are reduced, and normal blood pressure and cardiac output are maintained. In HF, however, a decrease in cardiac output leads to activation of the sympathetic nervous system and blunting of the baroreceptor reflex. The result is an elevation in heart rate, compensating for low cardiac output in an attempt to maintain perfusion to vital organs. As HF progresses, further depression of the baroreceptor function leads to greater sympathetic overactivity despite intense vasoconstriction and volume retention. Increasing activation of the sympathetic nervous system stimulates release of catecholamines from cardiac adrenergic nerves and the adrenal medulla; this in turn causes vasoconstriction in less metabolically active organs (e.g., skin, kidneys). It results in venoconstriction, which increases preload by increasing venous return. Catecholamines also affect the cardiac cells, producing an increased myocardial oxygen demand, myocyte hypertrophy, and tissue necrosis. Progressive HF occurs as cardiac cells progressively enlarge and die. As a result of sympathetic activation, plasma norepinephrine levels are elevated. The degree of plasma norepinephrine elevation correlates with the severity of HF and may be predictive of mortality, especially in patients with markedly elevated norepinephrine levels.6,7 Two additional vasoconstrictor systems act as compensatory mechanisms and therefore are affected by HF: the renin-angiotensin-aldosterone system and arginine vasopressin. The renin-angiotensin-aldosterone system is activated by a decline in blood pressure in the renal juxtaglomerular cells, which causes an increase in the release of the enzyme renin. The release of these hormones causes maladaptive remodeling of the ventricles; blockade of these hormones has been found to be beneficial in HF treatment.1 Cardiac remodeling is an alteration in both the structure and function of the heart as a response to cardiac injury or hemodynamic strain in association with neurohormonal activation. Remodeling can be physiologic (adaptive; as in the case of pregnant women and trained athletes) or pathologic (maladaptive; in disease states). Both myocardial hypertrophy and dilation occur in varying degrees as a progressive process, even in the absence of further myocardial injury, infection, or ischemic events.8 Dilation is an increase in the ventricular end-diastolic volume and represents an early compensatory response in volume overload in an attempt to increase contractility and to maintain cardiac output. In dilation, each individual myocyte lays down additional sarcomeres in series. Dilation preserves stroke volume and maintains cardiac output, but it also significantly increases wall stress, which increases myocardial oxygen demand, a deleterious condition if significant coronary artery disease is present. Also, excessive wall stress may lead to myocyte loss and fibrosis of cardiac tissue. The condition of ventricular hypertrophy is a direct result of attempts to compensate for the increase in wall stress. Ventricular hypertrophy is an increase in the number of sarcomeres within each myocyte of ventricular heart muscle; these abnormal, large cells cannot contract as efficiently. Initially, myocardial hypertrophy distributes the greater degree of wall stress to a greater myocardial mass and thus “normalizes” the increased load per myocyte. Hypertrophy also increases the force of the ventricular contraction. Ultimately, however, ventricular remodeling in HF progresses to the point that it can no longer offer any compensatory advantage, especially when loading conditions remain abnormal or when myocardial disease causes myocyte loss.9 At the onset of HF, compensatory mechanisms described are beneficial; however, over time, compensatory mechanisms may themselves exacerbate HF. The fluid retention intended to enhance contractile force can cause pulmonary and systemic congestion. Arterial vasoconstriction can cause impaired tissue perfusion and increased afterload. Myocardial hypertrophy and the sympathetic activity can increase myocardial oxygen consumption. The result of all these responses is an increase in myocardial burden and an escalation in frequency and severity of HF exacerbations. HF is a constellation of symptoms occurring as a result of heart muscle damage and the body’s physiologic response mechanisms. A careful history and review of symptoms are extremely important because they yield clues to the cause of HF; HF should never be the only diagnosis (Box 121-2). The cardinal symptoms of dyspnea and fatigue and the typical signs of HF, such as lower extremity edema and jugular venous distention, are nonspecific and must be evaluated along with a patient’s history, review of symptoms, and physical examination findings, corroborated by further cardiac investigations. Patients may describe symptoms of paroxysmal nocturnal dyspnea and orthopnea as well as shortness of breath with exertion or at rest. Some people experience abdominal fullness or bloating along with lack of appetite (Table 121-2). Certain signs, such as jugular venous distention, cardiac enlargement, and a third heart sound (S3), are specific for HF (70% to 90%), as is lower extremity edema, but these signs are only 11% to 55% sensitive (Table 121-3).10 TABLE 121-2 Symptoms of Heart Failure Symptoms and activity limitations have been quantified to assist in classifying a patient’s status. The American College of Cardiology (ACC) and the American Heart Association (AHA) have devised a classification system that grades HF by stage (Table 121-4) to include patients at risk for the development of HF (stage A) and those with end-stage, advanced disease (stage D). Complementary to the ACC/AHA staging system is the New York Heart Association (NYHA) classification, which illuminates the patient’s reported functional status and symptom burden. As an example, a patient with a prior history of HF who experiences shortness of breath while making the bed may be assigned to ACC/AHA stage C and NYHA class III HF.11 TABLE 121-4 Classification of Heart Failure
Heart Failure
Definition and Epidemiology
Parameter
HFrEF
HFpEF
HISTORY
Coronary artery disease
+++
++
Hypertension
++
++++
Diabetes
++
++
Valvular heart disease
++++
+
Paroxysmal dyspnea
++
+++
PHYSICAL EXAMINATION
Cardiomegaly
+++
+
Soft heart sounds
++++
+
S3 gallop
+++
+
S4 gallop
+
+++
Hypertension
++
++++
Mitral regurgitation murmur
+++
+
Rales
++
+
Edema
+++
+
Jugular venous distention
+++
+
CHEST X-RAY EXAMINATION
Cardiomegaly
+++
+
Pulmonary congestion
+++
+++
ELECTROCARDIOGRAPHY
Low voltage
+++
−
Left ventricular hypertrophy
++
++++
Q waves
++
+
ECHOCARDIOGRAPHY
Left atrial enlargement
++
++
Low ejection fraction
++++
−
Left ventricular dilation
++
−
Left ventricular hypertrophy
++
++++
Etiology
 Physician consultation is indicated for new onset of HF in a patient with no previous history of cardiac disease.
Physician consultation is indicated for new onset of HF in a patient with no previous history of cardiac disease.
 Physician consultation is recommended for patients with deterioration of previously stable HF.
Physician consultation is recommended for patients with deterioration of previously stable HF.
Cardiomyopathy.
Incidence, Prevalence, and Epidemiology
Risk Factors.
Pathophysiology
Systolic Dysfunction: Heart Failure with Reduced Ejection Fraction
Heart Failure with Preserved Ejection Fraction
Compensatory Mechanisms
Sympathetic Adrenergic Activity.
Neuroendocrine Activation.
Ventricular Remodeling.
Recurrent Acute Decompensated Heart Failure.
Clinical Presentation and Physical Examination
Symptoms
Why It Happens
What Patients with Heart Failure May Describe
Shortness of breath (dyspnea)
Pressure is increased in the pulmonary veins because the heart cannot keep up with the supply. This can cause pulmonary congestion or pulmonary edema (interstitial and alveolar congestion), which leads to left ventricular overload and worsening symptoms of failure.
Breathlessness during activity, at rest, or while sleeping (called paroxysmal nocturnal dyspnea); these symptoms worsen with severity of HF.
Difficulty breathing while lying flat (orthopnea) or complaints of waking up tired or feeling anxious and restless.
Persistent coughing, bronchospasm, or wheezing
Persistent pulmonary interstitial or alveolar edema (sometimes called cardiac asthma), worsen when recumbent.
Coughing that produces white or pink blood-tinged mucus may not always be present.
Edema
As blood flow out of the heart is impeded, blood returning to the heart through the veins backs up, causing fluid to build up in the tissues. The kidneys are less able to dispose of sodium and water, also causing fluid retention. This is evidence of right-sided HF.
Swelling in the feet, ankles, legs, or abdomen or weight gain.
Patients may find that their pants or shoes feel tight.
Tiredness, fatigue
The heart cannot pump enough blood to meet the needs of body tissues, resulting in decreased oxygen saturation. The body’s normal vascular response to exercise is altered; blood is diverted away from less vital organs, particularly muscles in the limbs, and sent to the heart and brain. Muscle deconditioning plays a role here as well. Loss of potassium induced by increased aldosterone also leads to muscle fatigue.
Tired feeling all the time and difficulty with everyday activities, such as shopping, climbing stairs, carrying groceries, or walking.
Lack of appetite, nausea
The digestive system receives less blood, causing problems with digestion. Medications, particularly diuretics, may be poorly absorbed. Hepatic congestion may often lead to discomfort.
Feeling of being full or nauseated, or loss of appetite.
Confusion, impaired thinking, lightheadedness
Changing levels of certain substances in the blood, such as sodium, can cause confusion. Poor cardiac output with decreased perfusion to the brain may also cause these symptoms.
Memory loss and feelings of disorientation; a caregiver or relative may notice this first.
Increased heart rate
Compensatory mechanism for the loss in stroke volume; the heart beats faster.
Heart palpitations, described by patients as a sensation that the heart is racing or throbbing.
Nocturia
Nocturnal diuresis lessens the degree of fluid retention. Nocturnal diuresis results from fluid reabsorption and redistribution in the supine position as well as a reduction in renal vasoconstriction that occurs at rest.
Patient reported; diuretics confound the picture.
Classification of Heart Failure
NEW YORK HEART ASSOCIATION FUNCTIONAL CLASSIFICATION
Class I
No limitations.
Ordinary physical activity does not cause undue fatigue, dyspnea, or palpitations.
Class II
Slight limitation of physical activity.
Such patients are comfortable at rest. Ordinary physical activity results in fatigue, palpitations, dyspnea, or angina.
Class III
Marked limitation of physical activity.
Although patients are comfortable at rest, less than ordinary activity will lead to symptoms.
Class IV
Inability to carry on any physical activity without discomfort.
Symptoms are present even at rest. With any physical activity, discomfort is experienced or increased.
ACC/AHA HEART FAILURE STAGES
Stage A
At high risk for HF with no identified structural or functional abnormality; no signs or symptoms.
Stage B
With structural heart disease that is strongly associated with the development of HF but without signs or symptoms.
Stage C
Symptomatic HF associated with underlying structural abnormalities.
Stage D
Advanced structural cardiac disease and marked symptoms of HF at rest with maximal medical therapy or requiring advanced therapies. 
Full access? Get Clinical Tree


Heart Failure
Chapter 121
 to
to  inch) in diameter. An impulse that is palpable with the palm of the hand, lasts longer, or is forceful indicates increased cardiac output or ventricular hypertrophy.
inch) in diameter. An impulse that is palpable with the palm of the hand, lasts longer, or is forceful indicates increased cardiac output or ventricular hypertrophy.