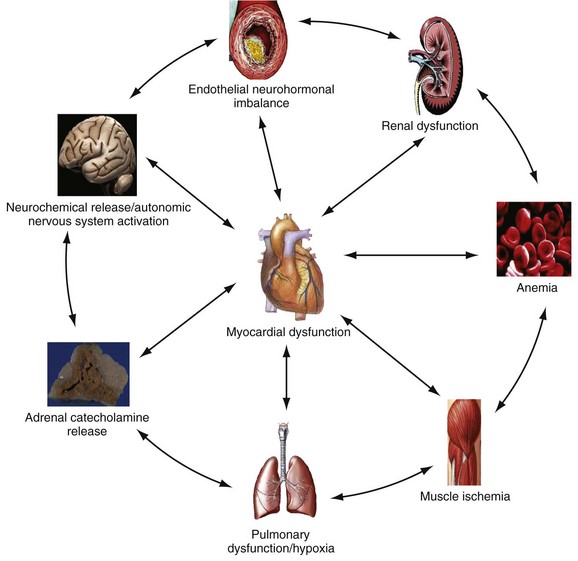
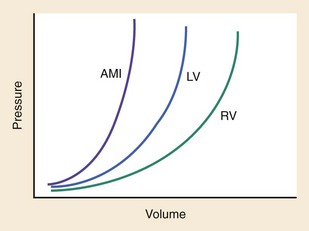
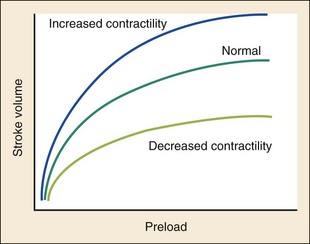
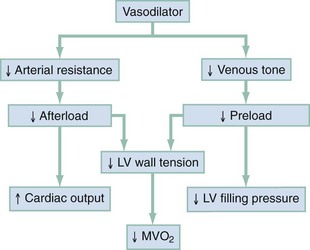
Chapter 81 HF is a progressive and multifaceted disease that begins long before symptoms and signs are evident. A complex neurohormonal regulatory relationship exists between the heart and multiple organ systems. Feedback loops mediated through a variety of vasoactive substances secreted by the heart, autonomic nervous system, kidneys, adrenals, lungs, and vascular endothelium are most important. Perturbations of function in any of these organs affect the others (Fig. 81-1). Accordingly, the cardiovascular system should be viewed as a dynamic one, continually adapting to optimize organ perfusion. Dysfunction of the heart or any component of the cardiopulmonary system initiates adaptive neurohormonal activation of the sympathetic nervous system, renin-angiotensin-aldosterone system (RAAS), natriuretic peptides, endothelin (ET), vasopressin, and other regulatory mechanisms. Neurohormonal activation initially compensates for circulatory system dysfunction. These mechanisms eventually lead to increased mechanical stress on the failing heart, however, causing maladaptive electrical and structural events, progressive cardiac fibrosis and apoptosis, and further impairment of systolic and diastolic function.1 This creates a vicious cycle of increasing myocardial dysfunction causing further neurohormonal modulation, leading to a progressive downward spiral. The degree of myocardial dysfunction depends on both the amount of primary myocardial disease and other pathologic conditions, particularly in the pulmonary, renal, and peripheral vascular systems. Understanding these underlying compensatory mechanisms is leading to progressive improvement in the management of HF, with a shift from a hemodynamic to a neurohormonal model. HF represents the only significant cardiovascular disease that is increasing in prevalence in our society. It is a common cause of poor quality of life and premature death. About 5,800,000 individuals (approximately 2% of the population) in the United States in 2009 had HF, and almost 550,000 new cases are diagnosed annually.2 The incidence approaches 10 per 1000 in people older than 65 years.3 Decompensated HF is the most common reason for hospital admission in this age group and also for readmission within 60 days of discharge.4 The ED is the main portal of entry for acute HF patients, with admission rate approximately 80%. HF results in an annual estimated health care cost of about $40 billion.5 It is also responsible for high rates of outpatient visits, hospitalizations, and readmissions. The aging population, coupled with improvements in the medical therapy of HF, will result in increased prevalence of this disease. HF carries approximately a 50% mortality at 5 years after symptom onset, and one third of patients with the most severe disease die within the first year after diagnosis.5,6 Females have a survival advantage over males.7,8 Progressive hemodynamic deterioration accounts for approximately 50% of deaths, but sudden death resulting from malignant ventricular dysrhythmias occurs in up to half.9 Multiple medical therapies decrease the death rate by improving functional status and slowing progression of pump dysfunction.10 The prognosis in HF is related to a number of factors, including age, LVEF,11,12 exercise tolerance, plasma norepinephrine and B-type natriuretic peptide (BNP) levels, cardiothoracic ratio on chest radiograph,13 resting heart rate (HR),14–16 electrocardiogram (ECG) evidence of left ventricular hypertrophy,17 atrial fibrillation (AF),18 or presence of ventricular dysrhythmias, hemoglobin A1c level,19,20 and renal function. One third to one half of patients with HF have some degree of renal insufficiency,21 which is one of the strongest predictors of mortality in patients with HF.22–24 HF disproportionately affects black compared with white Americans, and they have a higher mortality.25,26 Preload is the amount of force stretching the myofibril before contraction. In the intact ventricle, preload is produced by venous return into the chamber, resulting in stretch of the myofibrils constituting the chamber walls. The volume filling the chamber also results in development of pressure that can be measured in either ventricle. The pressure measured within a chamber is determined by both the volume stretching the chamber wall and compliance characteristics of the muscle. For this reason, ventricular pressure is only an indirect reflection of preload. Changes in compliance may occur acutely with ischemia or chronically with hypertrophy and may substantially alter the relationship between chamber volume, pressure, and preload (Fig. 81-2). Optimal preload is the filling pressure that stretches ventricular myofibrils maximally and leads to greatest stroke output per contraction. The actual optimal preload is unique for each patient because it is affected by LV loading conditions and compliance characteristics. For example, patients with acute myocardial infarction (AMI) tend to have a stiffer, less compliant left ventricle. In these patients, optimal LV pressure ranges are higher. Despite the inotropic state of the ventricle, optimizing preload results in the maximum stroke output for that ventricle (Fig. 81-3). Ventricles with normal compliance accommodate larger volumes before the chamber pressure rises. Accordingly, if pressure is used to estimate preload, the normal ventricle has more dramatic increases in stroke output for similar increases in filling pressure (steeper Starling curve). The risk of pulmonary edema increases when LV end-diastolic pressure rises significantly above normal ranges (6-12 mm Hg). In patients with low colloid osmotic pressures secondary to hypoalbuminemia, pulmonary edema may occur at even lower filling pressures. Afterload, for clinical purposes, can be thought of as the pressure against which the heart must pump to eject blood. Blood pressure (BP) is determined by the product of cardiac output (CO) and systemic vascular resistance (SVR) (BP = CO × SVR). Hypertension is a major contributor to HF in about 75% of cases.27 Patients with HF and low CO tend to maintain BP through peripheral vasoconstriction mediated mainly by endogenous catecholamines and the RAAS. Afterload represents the mural tension on myocardial cells during contraction and is determined by the total peripheral vascular resistance and the cardiac chamber size. The peripheral resistance is affected by the total cross-sectional area of the circulation, the blood viscosity, and other factors. The arterioles are the major resistance vessels in the circulation. Flow is directly proportional to the fourth power of the vessel radius (Poiseuille’s law). The larger the ventricular cavity, the more mural tension and thus myocardial work is required during contraction (law of Laplace). Failing ventricles have difficulty overcoming increases in peripheral resistance, instead dilating further, increasing end-diastolic volume to maintain SV, even with decreasing EF (preload reserve). Failing hearts are therefore extremely afterload sensitive, and modest vasodilation can dramatically increase CO, particularly in those most compromised (Fig. 81-4). Afterload reduction may be beneficial because it allows conversion of pressure work into flow work. When BP is decreased, CO increases as long as preload is maintained. Because flow work is proportionally less oxygen demanding, afterload reduction therapy has the additional benefit of decreasing myocardial oxygen demand. In developed countries, atherosclerotic coronary artery disease remains the leading cause of HF, present in almost 70% of patients in multicenter HF trials.28 Acute coronary thrombosis leads to focal myocardial necrosis, with resultant fibrosis and scarring. This process leads to areas of dyskinesis that result in decreased EF. Aneurysmal dilation of infarcted areas with paradoxical motion during systole may disproportionately decrease EF. When approximately 40% of the LV muscle mass is acutely infarcted, cardiogenic shock ensues. Transient loss of contractile function may result from episodes of myocardial ischemia that do not cause frank necrosis, or from an ischemic zone surrounding the infarct. This “myocardial stunning” may persist for several days. Owing to improved treatment of acute coronary syndromes, the rates of secondary death and HF are decreasing.29 Chronic coronary insufficiency leads to a more diffuse myocardial fibrosis termed ischemic cardiomyopathy. Revascularization of ischemic but not infarcted myocardial tissue provides a survival benefit in patients with HF related to ischemic LV systolic dysfunction.30 Diseases affecting the coronary microcirculation, such as vaso-occlusive sickle cell anemia and diabetes mellitus, result in similar pathology. Compensatory mechanisms may occur after large MI and progressive cardiac disease, which are collectively termed ventricular remodeling. They include cardiac dilation, reactive hypertrophy, progressive fibrosis, and changes in wall conformation. These may result from elevated chamber filling pressures as well as neurohumoral adaptive responses. Cardiomyopathies are a group of disease processes that primarily affect myocardium. Myocardial diseases resulting from coronary, valvular, and pericardial pathologies are excluded. Cardiomyopathy is categorized as primary if cause is unknown, or secondary if some cause is identified. Clinically, patients with cardiomyopathy tend to have three forms—dilated, hypertrophic, or restrictive—each associated with HF. Dilated cardiomyopathy is much more common than the other two and is the second most common cause of HF. The specific cardiomyopathies and myocarditis, which may also cause HF, are discussed in Chapter 82. Cardiac valvular disease is the third leading cause of HF, after ischemic heart disease and dilated cardiomyopathy. Most acute valvular dysfunction involves either the mitral or the aortic valve, and usually results in severe regurgitation. Acutely stenotic lesions are predominantly restricted to mechanical catastrophes of prosthetic valves. These patients may be in extremis with fulminant pulmonary edema. Valvular disease is discussed in Chapter 83. Pericardial diseases may significantly affect ventricular function, decreasing CO and increasing intracardiac pressures. In particular, cardiac tamponade may cause dyspnea and hypoperfusion not easily clinically distinguished from HF. ED use of bedside ultrasound has great usefulness in rapidly identifying this problem. Pericardial diseases are fully discussed in Chapter 82. Chronic obstructive pulmonary disease (COPD), which has a prevalence of 20 to 30% in HF, may obscure recognition of HF.31 Pulmonary dysfunction reduces myocardial oxygen supply while CO must be increased because tissue is being perfused with suboptimally oxygenated blood. Hypoxia leads to pulmonary arteriolar vasoconstriction, reducing lung vascular bed area and elevating pulmonary artery pressures. Chronic increases in pulmonary arterial pressure lead to right ventricular (RV) hypertrophy and dilation. When compensatory mechanisms fail, the patient develops right-sided HF (cor pulmonale), usually with LV output preserved, at least at rest. Causes of acute cor pulmonale, such as a large pulmonary embolus, may precipitate sudden systemic hypotension and death, partly because of decreased LV priming. Distinguishing primary pulmonary disease causing predominantly right-sided HF from LV failure with secondary right-sided dysfunction is clinically challenging. Wheezing or rhonchi may be seen in both entities. The chest radiograph may be difficult to interpret because both presentations cause interstitial changes. Hyperinflation depresses the diaphragm, which elongates the cardiac silhouette and may mask cardiomegaly. Competition for intrathoracic space reduces lung capacity in patients with chronic HF.32 Natriuretic peptide levels are only slightly elevated in primary pulmonary disease compared with much higher levels in LV failure.31,33,34 Initially, hypertrophy leads to improved function of each myocardial cell but at a higher energy cost. Hypertrophy is associated with myosin synthesis shifts from V1 to V3 isoforms, however, with related slowing of contraction, prolongation of time to peak tension, and reduced rate of relaxation.35 With the continued influence of volume overload, myofibril mass expands more than mitochondrial mass. Relative capillary blood flow is reduced, leading to progressive myocyte death with fibrosis and increased stress on the remaining myocytes. Thus the hypertrophic response, if allowed to continue, eventually becomes maladaptive, accelerating myocyte death and reducing pump function. This process gradually occurs as a normal part of aging.36 The heart and great vessels contain sensory receptors that detect changes in perfusion. Metabolic receptors in muscles also exert inhibitory and excitatory influences on brainstem vasomotor neurons. Arginine vasopressin (antidiuretic hormone) is released from the pituitary gland in response to decreases in perfusion. Elevated vasopressin levels in HF increase volume overload while decreasing osmolality. This adversely affects hemodynamics and cardiac remodeling while potentiating effects of angiotensin II and norepinephrine.37 Endothelial function locally regulates vasomotor tone. A family of ETs is produced by endothelial and smooth muscle cells as well as neural, renal, pulmonary, and inflammatory cells. This occurs in response to hemodynamic stress, hypoxia, catecholamines, angiotensin II, and many inflammatory cytokines. ET-1 is the most important ET and the most potent vasoconstrictor known.38 It exerts its main vascular effects, vasoconstriction and cell proliferation, through specific ETA and ETB receptors on vascular smooth muscle cells. ETA receptor activation causes vasoconstriction, whereas ETB receptor stimulation increases prostacyclin and nitric oxide (NO) release, causing vasodilation. ET-1 plasma levels are elevated in HF, correlate with symptoms as well as hemodynamic stress, and are associated with adverse prognosis. NO is produced in almost all tissues and plays a critical role in the homeostasis of cardiac function.39 NO exerts its biologic signaling through production of cyclic guanosine monophosphate (cGMP), which is broken down by cyclic nucleotide phosphodiesterases (PDEs).40 Reduced synthesis or increased degradation of NO at the endothelial level is detrimental in HF. NO-mediated endothelial dysfunction may represent the earliest stage of target organ damage, which ultimately leads to hypertensive heart disease and HF.41 The prototypical case of acute HF involves a healthy person who develops a large myocardial infarction (MI) or acute valvular dysfunction. Chronic HF is best characterized by a disease state such as dilated cardiomyopathy, with gradual deterioration of cardiac function. In acute HF, early presentation may be a result of systolic dysfunction and hypoperfusion, often with acute pulmonary edema (APE) resulting from sudden reduction in chamber compliance. Chronic HF usually arises with symptoms related to fluid retention, with compensatory mechanisms adjusted so that normal perfusion exists, at least in the resting state. In clinical practice, approximately 80% of HF cases seen the ED involve acute decompensation of chronic heart disease.42 Systolic dysfunction refers to impairment of contractility, with stroke output reduced and forward flow compromised. Systolic dysfunction is typically caused by myocyte damage such as in MI or myocarditis. Asymptomatic LV systolic dysfunction in patients 45 years of age or older has an estimated prevalence of 6% and is much more common than symptomatic systolic HF.43,44 Most cases of systolic dysfunction also involve some degree of diastolic dysfunction. Diastolic dysfunction indicates a primary problem with ability of the ventricles to relax and fill normally.45 In many cases, normal or even supernormal systolic function is preserved. Echocardiographic and nuclear imaging techniques demonstrate that 40 to 50% of patients with congestive symptoms have normal EFs and experience diastolic dysfunction,46,47 also termed heart failure with normal ejection fraction.48 The proportion of HF that is primarily diastolic increases with age, from about 45% in patients younger than 45 years to almost 60% in patients older than 85 years. Asymptomatic diastolic dysfunction is much more common than asymptomatic systolic dysfunction. Diastolic dysfunction is the predominant pathophysiology in hypertrophic and restrictive cardiomyopathies, valvular aortic stenosis, and, most important, hypertension. Diastolic dysfunction occurs predominantly as a result of one of three mechanisms: impaired ventricular relaxation, increased ventricular wall thickness, or accumulation of myocardial interstitial collagen. Impaired lusitropic capacity of the myocardium leads to higher ventricular filling pressure, resulting in congestive symptoms. Myocardial relaxation is an active, energy-requiring process. Failure of myocytes to relax may be secondary to low intracellular energy stores. Physiologic stresses causing increased cardiac demands can precipitate lusitropic abnormalities. In chronic renal disease, mortality is higher in diastolic than systolic HF.49 In addition, systolic contractile dyssynchrony occurs in one third of diastolic HF patients, whereas diastolic dyssynchrony is present in more than half, with therapeutic implications.50,51 As with the other classification schemes, most patients with HF have components of both systolic and diastolic dysfunction, with the predominant type allowing specific treatment strategies. For example, patients with predominantly diastolic dysfunction have the advantage of intact myocardial contractile function. Stiffer hearts, however, have steep pressure-volume curves. Therefore small reductions in diastolic filling volume, as may occur with aggressive vasodilator or diuretic therapy, may markedly decrease ventricular filling and compromise stroke output (see Fig. 81-3). Precipitating Causes of Heart Failure and Exacerbating Factors When cardiac decompensation occurs because of an acute precipitating cause, intervention can focus on the precipitating factor(s), and the prognosis is much better than when there is simply progression of intrinsic cardiac failure. Causes of acute cardiac decompensation are provided in Box 81-1. Sudden elevation of arterial pressure acutely increases afterload, which may precipitate rapid onset of HF. This is particularly common when antihypertensive therapy is abruptly discontinued. Pheochromocytoma and other states associated with high sympathetic outflow may be implicated. Cocaine and other sympathomimetic drugs of abuse frequently precipitate HF. Emotional upset can dramatically increase afterload as well as precipitate coronary vasospasm, with the extreme example being acute stress cardiomyopathy (Takotsubo syndrome; see Chapter 78). A new ischemic event may precipitate HF by impairing contractility and decreasing LV compliance. Pulmonary edema may occur rapidly in this setting, especially when large areas of myocardium are involved. In the compromised heart, even local ischemia may precipitate HF. Occult acute coronary syndrome is common, particularly in the elderly, and should be considered in any HF exacerbation. The presence of HF on admission in patients with acute coronary syndromes is associated with increased short- and long-term rates of death and MI.52–54 Infection results in increased systemic metabolic demands. The sepsis syndrome is associated with a reversible form of myocardial depression, mediated by various cytokines, including interleukin (IL)-1, IL-2, and IL-6, as well as tumor necrosis factor.55 Proinflammatory cytokines such as tumor necrosis factor alpha and IL-6 also play an important pathogenic role in chronic HF.56 The prevalence of AF in patients with HF increases from less than 10% in New York Heart Association (NYHA) functional class I to approximately 50% in NYHA functional class IV.57 Neurohormonal alterations, electrophysiologic changes, and mechanical factors create an environment in which HF predisposes to AF and AF exacerbates HF. New-onset AF or other dysrhythmias that affect coordinated atrial priming of the ventricular pump may seriously reduce preload, especially in disease states with reduced ventricular compliance. Significant bradydysrhythmias may also reduce CO simply by reducing the number of systolic ejections per minute (CO = SV × HR). Both COPD exacerbations and respiratory tract infections are very important precipitating factors for HF exacerbation.58 Pulmonary infection, which is more common in patients with pulmonary vascular congestion, may add hypoxia to the metabolic stressors of fever, tachycardia, and increased tissue perfusion requirements. With chronic anemia, oxygen delivery to tissues is maintained by increased CO (isovolumic hemodilution). Anemia increases in prevalence with increasing severity of HF, especially with declining renal function and increasing age. Anemia is associated with poorer survival in HF,59–61 with greater disease severity, greater LV mass index, and higher hospitalization rates. Abrupt exacerbations of anemia increase systemic perfusion demands and, especially if coupled with reduced coronary oxygen delivery, may prompt onset or exacerbation of HF. HF may be a clinical manifestation in patients with previously compensated cardiac disease who develop hyperthyroidism. Hypothyroidism also adversely affects myocardial pump function. Restoration of normal thyroid function usually reverses the abnormal cardiovascular hemodynamics.62 Beta-blocking and calcium channel blocking agents have negative inotropic effects and may precipitate overt HF with excessive administration. Many antidysrhythmic agents have similar effects. Glucocorticoids, NSAIDs, vasodilator medications, and others may result in sodium retention with substantial increases in plasma volume that may precipitate HF.63 NSAIDs in particular interfere with prostaglandin synthesis through cyclooxygenase inhibition, thereby impairing renal homeostasis in patients with HF. They also interfere with the effects of diuretics and ACE inhibitors. Nonadherence to medication regimens for hypertension, HF, or ischemia is the most common pharmacologic cause of HF decompensation. The NYHA classification system is a time-honored categorization for patients with chronic HF based on degree of activity causing symptoms (Box 81-2).64 Careful consideration of the differential diagnosis of HF is symptom based. The most common manifestation of acute HF is respiratory distress caused by pulmonary edema. Therefore the differential diagnosis includes exacerbation of COPD or asthma, pulmonary embolus, pneumonia, anaphylaxis, and other causes of acute respiratory distress. Hypoperfusion may be caused by some of these as well as by sepsis syndrome, hypovolemia, hemorrhage, cardiac tamponade, and tension pneumothorax. Paroxysmal nocturnal dyspnea results from pulmonary congestion precipitated by plasma volume expansion that occurs during recumbency as interstitial edema is reabsorbed into the circulation. Orthopnea occurs through the same mechanism, with the supine position causing rapid increases in diastolic filling pressure. Symptoms abate after the patient stands or props up the trunk and venous return decreases. Nocturia results from the same pathophysiologic process. Many historical features increase the likelihood of HF. Most predictive is a past history of HF or paroxysmal nocturnal dyspnea, and the absence of dyspnea on exertion reduces the likelihood of chronic HF.65 Most HF patients are hypertensive, which is prognostically preferable to normal or low BP. Clammy, vasoconstricted patients with a thready pulse and delayed capillary refill may have systemic hypoperfusion despite adequate BP, which is maintained by intense vasoconstriction. Noninvasive assessment of BP in the vasoconstricted patient with low CO can be inaccurate.66 If available, intra-arterial pressure monitoring may more accurately reflect the systemic hemodynamic state and guide the choice of inotropic or vasoconstrictor therapy in hypotensive HF patients. These common clinical findings of chronic HF are prevalent among patients with APE because most patients have acute exacerbations superimposed on chronic underlying disease. Jugular venous distention, pedal edema, and cardiomegaly may be absent in previously healthy individuals with pulmonary edema resulting from an initial episode of acute HF. The presence of a third heart sound significantly increases the likelihood of HF, whereas absence of rales decreases the likelihood.65 Physical examination of patients with APE resulting from acute MI may identify surgically correctable lesions such as acute mitral regurgitation or ventricular septal defect. An upright chest radiograph helps distinguish cardiogenic pulmonary edema from other causes of dyspnea. An enlarged cardiac silhouette is seen in 70% of cases. A normal heart size suggests acute cardiogenic pulmonary edema in a patient without prior HF, diastolic dysfunction, COPD, or noncardiogenic pulmonary edema. An early ECG for arrhythmia recognition and management is important, as well as for identification of acute coronary syndrome. Absence of cardiomegaly on chest radiography and a normal ECG greatly decrease the likelihood that HF is causing the presentation.65 Obtaining a complete blood count (CBC) to evaluate for anemia and a basic metabolic panel to determine electrolyte status as well as renal function is generally useful. Cardiac biomarkers help evaluate for ongoing myocyte injury, which may be clinically silent. In most cases, and particularly when the diagnosis of HF is unclear, natriuretic peptide levels should be obtained. Pre-proBNP is synthesized in the ventricles in response to myocyte stretch, then released and enzymatically cleaved to NH2-terminal–proBNP (NT-proBNP) and BNP. NT-proBNP and BNP levels help identify patients with HF and may improve management of patients in the ED with dyspnea.67–69 The “breathing not properly” BNP Multinational Study was a prospective evaluation of patients who came to the ED with acute dyspnea. BNP levels above 500 pg/mL were highly associated with HF (likelihood ratio [LR] 8.1), and levels of 100 to 500 pg/mL were generally indeterminate (LR = 1.8). A low BNP level (<100 pg/mL) indicated that HF was highly unlikely (LR = 0.13).70,71 ED use of BNP and NT-proBNP assays aids diagnosis of HF and can reduce admission rates and length of hospitalization in acute dyspnea.72 Natriuretic peptide levels correlate with ventricular function, NYHA classification, and prognosis.73–76 Results of large clinical trials confirm that BNP levels are the strongest predictor of outcome in HF compared with other neurohormones and clinical markers.77 There is often a disconnect between the perceived severity of HF by clinicians and the degree of BNP elevation, yet BNP levels are better predictors of 90-day outcome than physician judgment.78 High predischarge BNP and NT-proBNP levels are strong, independent predictors of death or rehospitalization after decompensated HF.79,80 NT-proBNP– and BNP-guided therapy reduces all-cause mortality and provides a strong measure of therapeutic response in chronic HF compared with usual care.81–83 Mildly elevated BNP levels may also be seen in right-sided HF related to cor pulmonale or pulmonary embolism. BNP levels are only slightly elevated in patients with end-stage renal disease, and in this setting marked elevation reflects ventricular dysfunction.84 Admission BNP and troponin levels are independent predictors of in-hospital mortality in acute decompensated HF.85 Increased concentrations of these and other biochemical markers of myocyte injury in the absence of discrete ischemic events in HF help identify patients most likely to have adverse outcomes.86 Plasma levels of cardiac troponins in stable HF also predict adverse outcomes.87,88 The ESCAPE trial demonstrates that pulmonary artery catheterization in severe symptomatic HF increases anticipated adverse effects but does not affect overall mortality or duration of hospitalization.89 Noninvasive impedance cardiography appears to be an effective and developing technology to measure CO and other hemodynamic variables in HF and may obviate the need for a pulmonary artery catheter, although it cannot reliably measure LV filling pressures.90,91 Bedside ultrasound can be an important ED screening tool in HF, with attention to wall motion abnormalities, EF, and valvular function, as well as exclusion of cardiac tamponade. Echocardiography is similarly useful and can provide detailed measures of LV function and determine structural heart disease.92,93 Multidetector computed tomography coronary angiography can distinguish ischemic from other forms of cardiomyopathy but is rarely useful in acute HF.94 Radionuclide imaging and cardiac magnetic resonance imaging have an expanding role in evaluating chronic HF but no utility in the acute setting. Of patients with HF in the ED, about 20% are experiencing their first episode of HF, and 80% have had prior hospital visits for the same condition.42 The approach focuses on (1) determining underlying cardiac pathology, (2) identifying the acute precipitant, and (3) mitigating the acute decompensation. The immediate therapeutic goals are to improve respiratory gas exchange, maintain adequate arterial saturation, and decrease LV diastolic pressure while maintaining adequate cardiac and systemic perfusion. Many patients with acute HF demonstrate adequate systemic perfusion with elevated BP because of activation of various compensatory mechanisms. The ability of the left ventricle to generate normal or elevated systolic pressures indicates the presence of considerable myocardial reserve and is associated with lower mortality in both acute and chronic HF.95 These patients should be quickly distinguished from those with pulmonary edema and evidence of hypoperfusion. Hypertensive pulmonary edema is easier to manage because afterload reduction with vasodilators is extremely effective. Therapeutic interventions should decrease both preload and afterload. Excessive preload reduction may result in an abrupt decrease in CO, however, which could cause hypotension. This occurs more readily in patients with less compliant hearts. Fluid challenge generally restores BP. Therapy for APE with adequate perfusion should begin with upright positioning, supplemental oxygen, nitrates, morphine sulfate, and loop diuretics (Fig. 81-5). This allows prompt improvement in most of these patients.
Heart Failure
Epidemiology
Cardiac Physiology
Primary Disease Processes Resulting in Heart Failure
Coronary Artery Disease
Cardiomyopathy and Myocarditis
Valvular Heart Disease
Pericardial Diseases
Pulmonary Disease
Compensatory Mechanisms in Heart Failure
Development of Cardiac Hypertrophy
Neurohormonal Modulation
Central and Autonomic Nervous System Neurohormonal Response
Vascular Endothelial Neurohormonal Response
Classification of Heart Failure
Acute versus Chronic Heart Failure
Systolic versus Diastolic Dysfunction
Clinical Evaluation of Patients with Suspected Heart Failure
Systemic Hypertension
Myocardial Infarction and Ischemia
Systemic Infection
Dysrhythmia
Acute Hypoxia and Respiratory Problems
Anemia
Thyroid Disorders
Pharmacologic Complications
Evaluation of Heart Failure
History
Physical Examination
Diagnostic Testing in Heart Failure
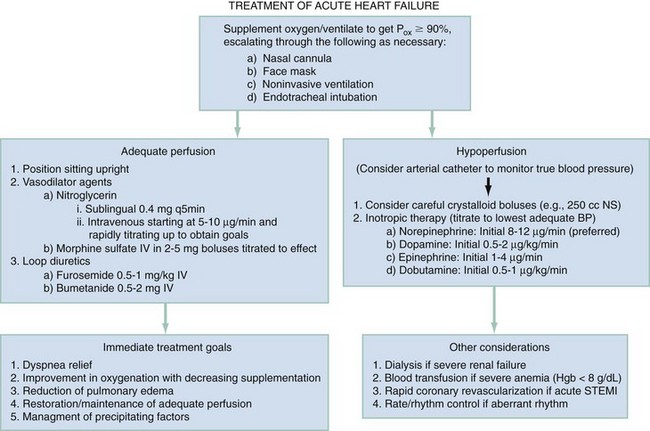
Treatment of Acute Heart Failure
Acute Heart Failure with Adequate Perfusion
< div class='tao-gold-member'>

Full access? Get Clinical Tree


Heart Failure
Only gold members can continue reading. Log In or Register to continue




