CHAPTER 10
Heart Failure
Judy Cheng, PharmD, MPH, BCPS, FCCP • Jason T. Slyer, DNP, RN, FNP-BC, CHFN, FNAP
Heart failure is a public health problem of enormous and growing significance. It affects more than 5 million people in the United States. As the population ages and the incidence of patients surviving other symptomatic cardiac diseases such as myocardial infarction (MI) continues to rise, the incidence of heart failure and its mortality rate will continue to increase. Heart failure is the only cardiovascular disease that is increasing in prevalence.
The management of heart failure has become one of the most challenging problems confronting our health care system today. Each year, the United States spends nearly $32 billion in managing patients with heart failure, with projected increases of 120% to $70 billion per year by 2030 (Go et al., 2013).
Treatment of heart failure is no longer confined to symptom relief. Because the underlying etiologies that contribute to ventricular dysfunction may progress independently from the development of symptoms, treatment to prevent and delay the progression of the disease is equally important. This chapter discusses the pathophysiology and treatment of heart failure, with emphasis on long-term management and patient education to prevent exacerbation and delay the disease progression to improve a patient’s quality of life.
 ANATOMY, PHYSIOLOGY, AND PATHOLOGY
ANATOMY, PHYSIOLOGY, AND PATHOLOGY
Functional Anatomy of the Heart
The heart is a four-chambered structure consisting of two atria and two ventricles. The atria are relatively thin-walled, low-pressure chambers that lie superior to the ventricles. Their primary function is to act as a reservoir, filling their respective ventricles. The ventricles are thick-walled chambers that function at higher pressures and pump blood from the heart into the respective great vessels. The atria and ventricles are more specifically identified as either right or left, according to their orientation in the chest. The right atrium and ventricle receive deoxygenated blood as it returns from the body. The right ventricle then pumps this blood to the lungs through the pulmonary artery. After the blood is oxygenated in the lungs, it returns via the pulmonary veins to the left atrium and is then pumped out to the rest of the body by the left ventricle via the aorta. The circulatory system thus consists of two circuits in series, the pulmonary and systemic, through which the blood sequentially flows.
The unidirectional blood flow is maintained by the four valves located in the heart. These valves allow the forward flow of blood and when closed prevent retrograde flow. The two atrioventricular valves, mitral and tricuspid, in the right and the left sides of the heart respectively, separate the ventricles from their respective atria. The semilunar valves, aortic and pulmonic, divide the ventricles from their respective great vessel. The left ventricle is separated from the aorta by the aortic valve; the right ventricle is separated from the pulmonary artery by the pulmonic valve.
Cardiac Physiology and Hemodynamics
The primary physiological function of the heart is to pump blood to supply oxygen and nutrients to the different body organ systems. Stroke volume is the volume of blood pumped per beat by each ventricle. Cardiac output is the volume of blood pumped per minute. Cardiac output and stroke volume are therefore related by the following equation:
![]()
Stroke volume is regulated by three variables: the end-diastolic volume (EDV), the mean aortic or arterial blood pressure, and the contractility of the ventricles. The EDV is the amount of blood in the ventricles just before contraction. Because this is a workload imposed on the ventricles before contracting, it is clinically referred to as the preload. Arterial pressure represents an impedance to the ejection of blood from the ventricles, or an afterload imposed on the ventricles after contraction has begun. The stroke volume is directly proportional to the preload and contractility but inversely proportional to the afterload.
The portion of the EDV that is ejected (the ejection fraction or EF) against a given afterload depends on the strength of ventricular contraction. Normally, contraction strength is sufficient to eject two thirds of the EDV with each heart beat. In a healthy heart, this fraction remains relatively constant, even with an increase in EDV. This implies that the strength of ventricular contraction must increase as the EDV increases in a normal heart. Such intrinsic control of contractile strength was first described by two physiologists, Frank and Starling, and thus is named the Frank–Starling law of the heart.
The other factor that controls cardiac output is the heart rate. Heart rate is regulated by the autonomic nervous system. Stimulation of the sympathetic nervous endings in the musculature of the atria and ventricles increases the strength of contraction (positive inotropic effect) and decreases slightly the time spent in systole. In contrast, enhancing the effect of the parasympathetic nervous system decreases cardiac rate and contractility.
Pathophysiology of Heart Failure
The pathophysiology of heart failure begins with myocardial cell damage caused by etiologies such as ischemic heart disease and hypertension. Table 10.1 details the causes of heart failure (Yancy et al., 2013). As myocardial damage becomes significant, either myocardial contractility decreases or the left ventricle stiffens and is unable to fill to full capacity. Both of these conditions lead to a decrease in cardiac output and intraventricular pressure increases to the point where body organ perfusion and functions are compromised. In the left ventricle, an increase in diastolic pressure leads to a rise in pressure in the pulmonary circulation. This causes pulmonary edema and prevents proper oxygenation of the blood, leading to dyspnea. In the right ventricle, an increase in diastolic pressure leads to an elevation of venous pressure, resulting in peripheral edema.
Etiologies of Heart Failure |
 Ischemic cardiomyopathy
Ischemic cardiomyopathy
 Coronary artery disease
Coronary artery disease
 Nonischemic, dilated cardiomyopathy
Nonischemic, dilated cardiomyopathy
 Hypertension
Hypertension
 Valvular heart disease
Valvular heart disease
 Infectious etiologies
Infectious etiologies
 Toxic cardiomyopathy
Toxic cardiomyopathy
– Alcohol
– Cocaine
– Cardiotoxicity related to chemotherapy (e.g., doxorubicin)
– Other cardiotoxins (e.g., ephedra, anabolic steroids)
 Tachycardia induced
Tachycardia induced
 Cardiomyopathy from inflammation
Cardiomyopathy from inflammation
– Rheumatologic disorders (e.g., systemic lupus erythematosus, scleroderma)
– Peripartum cardiomyopathy
– Iron overload
– Amyloidosis
– Cardiac sarcoidosis
 Genetic defects/familial cardiomyopathy
Genetic defects/familial cardiomyopathy
 Obesity
Obesity
 Diabetes mellitus
Diabetes mellitus
In the presence of a primary abnormality in myocardial contractility or excessive hemodynamic stresses, the heart relies on three major adaptive mechanisms in attempts to maintain cardiac output:
 The Frank–Starling mechanism, in which an increase in preload brought about in part by salt and water retention helps sustain cardiac performance (stroke volume is directly proportional to preload)
The Frank–Starling mechanism, in which an increase in preload brought about in part by salt and water retention helps sustain cardiac performance (stroke volume is directly proportional to preload)
 Increased release of catecholamines by adrenergic cardiac nerves and the adrenal medulla activation of the renin–angiotensin–aldosterone system, and other neurohormonal adjustments that act to maintain arterial pressure and vital organ perfusion
Increased release of catecholamines by adrenergic cardiac nerves and the adrenal medulla activation of the renin–angiotensin–aldosterone system, and other neurohormonal adjustments that act to maintain arterial pressure and vital organ perfusion
 Myocyte hypertrophy with or without chamber dilatation, in which left ventricular mass is increased in an attempt to enhance contractility
Myocyte hypertrophy with or without chamber dilatation, in which left ventricular mass is increased in an attempt to enhance contractility
Figure 10.1 illustrates the interactions of the different compensatory mechanisms in heart failure.
Initially, these mechanisms can maintain cardiac output, arterial blood pressure, and organ perfusion, but they may also exert more stress on the already injured myocardium. Therefore, in the later phase of the disease, such compensatory mechanisms actually contribute to the worsening of symptoms. Table 10.2 illustrates the short-term and the long-term responses of the impaired myocardium to these compensatory mechanisms.
Hemodynamic Compensatory Mechanisms in Heart Failure
In the early phase of heart failure, activation of the sympathetic nervous system increases both the heart rate and the contractile force, thus increasing cardiac output. However, an important cardiac compensatory mechanism called myocardial remodeling, which occurs during myocardial injury and in a state of enhanced sympathetic nervous activities, may be both adaptive and maladaptive (Francis, 2001; Zipes, Libby, Bonow, & Braunwald, 2005). Unlike skeletal muscle cells, myocardial cells cannot divide to increase their numbers, but they undergo remodeling by which they increase in length or volume (dilatation and hypertrophy). Early in heart failure, dilatation and hypertrophy may enhance contraction, but chronically they often worsen cardiac damage. In addition, dilatation of heart chambers leads to an increase in myocardial wall stress, which is one of the determinants of myocardial oxygen demand and supply imbalance. Hypertrophy, if severe and long-standing, leads to loss of contractile force.
Neurohormonal Compensatory Mechanisms in Heart Failure
One of the most significant compensatory mechanisms of heart failure is the activation of certain endogenous neurohormones. Reduction of cardiac output stimulates other body systems to try to maintain normal blood pressure and organ perfusion. The major neurohormonal systems that regulate these compensatory mechanisms are the sympathetic nervous system, the renin–angiotensin–aldosterone system, and the natriuretic peptides.
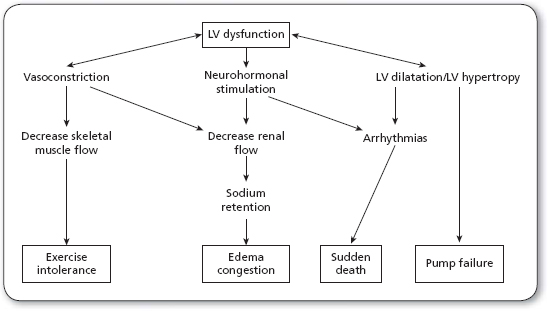
FIGURE 10.1
Pathophysiology of heart failure.
LV, left ventricular.
Short-Term and Long-Term Responses of Impaired Myocardium to Different Compensatory Mechanisms | ||
| SHORT-TERM EFFECTS | LONG-TERM EFFECTS |
Frank–Starling mechanisms: salt and water retention (renin–angiotensin activation) | Increases preload | Causes peripheral and pulmonary congestion |
Sympathetic nervous stimulation | Maintains blood pressure and organ perfusion by vasoconstriction | Increases energy expenditure; long-term stimulation also leads to desensitization of adrenergic receptors |
Myocardial hypertrophy | May enhance contraction | Deterioration and death of cardiac cells |
SYMPATHETIC NERVOUS SYSTEM
Plasma norepinephrine levels are increased in heart failure (Latini et al., 2004). With such activation of the sympathetic nervous system, there is an initial increase in cardiac contractility. Vasoconstriction also occurs and afterload increases. As mentioned, this is intended to enhance cardiac output and maintain vital organ perfusion. Chronically, however, this leads to an increase in systemic vascular resistance and adds to the strain of a failing heart. Increasing afterload leads to a decrease in forward blood flow to perfuse the body’s organ systems. Decreased forward flow in turn results in a backup of venous blood returning to the heart, or increased preload. Vasoconstriction also leads to a reduction of blood flow to the kidneys, which stimulates the retention of sodium and water to compensate for the perceived lack of blood volume. Such retention of fluid and water does not improve stoke volume; rather, it contributes to the congestive symptoms of heart failure.
RENIN–ANGIOTENSIN SYSTEM
In states of low cardiac output, the renin–angiotensin–aldosterone system is activated (Ma, Kam, Yan, & Lam, 2010). This acts in concert with the activated adrenergic nervousadrenal medullary system to maintain arterial pressure. Renin is released by the kidneys in response to reduced blood flow. Renin converts angiotensinogen to angiotensin I in the circulation. Angiotensin I circulates to the lungs and other tissues, where angiotensin-converting enzyme (ACE) converts it to angiotensin II. Angiotensin II is a potent vasoconstrictor and therefore significantly increases afterload. Aldosterone production is also increased by angiotensin II. Aldosterone has potent sodium-retaining properties and contributes to the general volume overload state of heart failure. Finally, angiotensin II plays an important role in the stimulation of the cell growth and development that leads to myocyte hypertrophy of the heart (Figure 10.2).
NATRIURETIC PEPTIDES
Atrial natriuretic peptide (ANP) is produced by the atrial tissue of the heart. Brain natriuretic peptide (BNP) is produced by the ventricular tissue. Secretion of ANP and BNP is regulated by wall tension; therefore, their levels are increased in chronic heart failure (Zipes et al., 2005). ANP and BNP are counterregulatory hormones that oppose the action of many of the vasoconstricting and salt- and water-retaining effects of the renin–angiotensin–aldosterone system. ANP and BNP act as vasodilating agents, suppress the formation of renin, and enhance the excretion of salt and water. However, because of their relatively weak action and short duration of effect, they cannot totally reverse the detrimental effects of the renin–angiotensin–aldosterone system and the sympathetic systems.
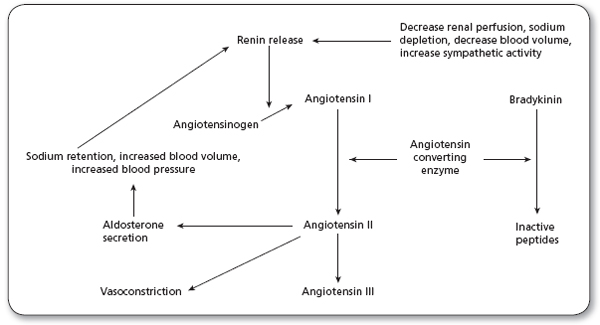
FIGURE 10.2
The renin–angiotensin–aldosterone system.
 EPIDEMIOLOGY
EPIDEMIOLOGY
More than 5 million Americans suffer from heart failure and more than 650,000 new cases are diagnosed each year (Go et al., 2013). Despite recent advances in the management of heart failure, both surgical and pharmacological, the mortality rate of this disease remains high. The 5-year mortality rate is approximately 50%. The increasing average age of the American population and the longer survival of people with other cardiac and comorbid diseases who subsequently develop heart failure at an older age add to the rapid increase in heart failure prevalence. Race-specific differences reflected a higher incidence of heart failure for African Americans, followed by Hispanic, White, and Chinese Americans. The risk of developing heart failure before age 50 years is higher in African Americans than Whites. This increased risk in African Americans is reflective of difference in risk factors such as hypertension, obesity, and diabetes mellitus, as well as differences in socioeconomic status (Go et al., 2013).
The management of heart failure patients exerts a heavy economic burden on society. Annually, more than 1 million patients are admitted to the hospital for heart failure management, accounting for a total Medicare expenditure exceeding $17 billion annually. Mortality and readmission rates following a heart failure hospitalization remain high, with nearly 1 in 4 patients readmitted to hospital within 30 days of discharge. As prevention of hospitalization and readmission has become a national priority, there is a growing fiscal imperative to develop strategies to improve the transition from hospital to home and provide more effective ambulatory heart failure management.
 DIAGNOSTIC CRITERIA
DIAGNOSTIC CRITERIA
Heart failure is characterized by a pathophysiological state in which the heart cannot provide adequate forward cardiac output to meet the perfusion and oxygenation requirements of the body organs and tissues. Although the etiologies of heart failure are numerous, the majority of cases can be classified as either heart failure with reduced ejection fraction (HFrEF), also referred to as systolic heart failure, in which there is impaired cardiac contractility; or heart failure with preserved ejection fraction (HFpEF), also referred to as diastolic heart failure, in which decreased compliance of the heart (manifesting as the heart’s inability to relax) impairs ventricular filling. Distinguishing between these disorders is important clinically because they are managed differently. Nevertheless, there is common symptomatology, including fatigue, shortness of breath at rest, dyspnea on exertion, peripheral/pulmonary edema, and weight gain caused by fluid retention and congestion.
HFrEF is characterized by a dilated left ventricular chamber with a poorly contracting left ventricle and usually a thinned ventricular wall, ultimately producing a reduced left ventricular EF (≤40%; Yancy et al., 2013). In patients with HFpEF, the left ventricular chamber size may not increase and the wall thickness is usually enhanced. The most important diagnostic criterion for distinguishing HFrEF and HFpEF is the left ventricular EF. Patients with HFpEF may have a normal or increased left ventricular EF. Table 10.3 summarizes the major characteristics and diagnostic criteria of HFrEF and HFpEF.
Diagnostic Criteria of Heart Failure | ||
| HEART FAILURE WITH REDUCED EJECTION FRACTION (HFrEF) | HEART FAILURE WITH PRESERVED EJECTION FRACTION (HFpEF) |
Symptoms | Fatigue, shortness of breath, dyspnea on exertion, pulmonary or peripheral congestion, fluid retention | Similar to HFrEF |
Left ventricular ejection fraction | ≤40% | ≥50% |
Heart chamber size | Increased | Normal or increased |
Wall thickness | Thinned | Typically enhanced |
Medications That May Induce or Exacerbate Heart Failure |
MEDICATIONS | EFFECTS |
Antiarrhythmic agents (e.g., quinidine, procainamide, flecainide, propafenone, sotalol) | Most antiarrhythmic agents have negative inotropic effects and may induce heart failure when used in patients with other underlying heart diseases |
Non-dihydropyridine calcium channel blockers (verapamil, diltiazem) | Most first-generation, non-dihydropyridine calcium channel blockers have negative inotropic effects and may induce heart failure when used in patients with other underlying heart diseases. Second-generation, dihydropyridine-derivative calcium channel blockers (amlodipine and felodipine) may be considered for hypertension management, but they have not demonstrated any benefit in heart failure symptoms or survival |
Heroin, cocaine, alcohol, amphetamines, doxorubicin, cyclophosphamide, sulfonamides, lead, arsenic, cobalt, phosphorus, ethylene glycol | Direct cardiac toxins |
Corticosteroid and nonsteroidal anti-inflammatory agents (NSAIDs) | Cause salt and water retention |
Thiazolidinedione (e.g., pioglitazone) | Causes plasma volume expansion |
 HISTORY AND PHYSICAL EXAMINATION
HISTORY AND PHYSICAL EXAMINATION
Obtaining a complete medical history is extremely important in diagnosing heart failure. Identifying other health conditions or behaviors that may accelerate the progression of heart failure is necessary for effective management. Patients should be questioned about a previous history of angina or the equivalent (such as flash pulmonary edema); MI; hypertension; other heart diseases; diabetes; and renal, pulmonary, thyroid, or gastrointestinal diseases. A complete medication history is also important to obtain. Table 10.4 lists some common medications that may cause or exacerbate heart failure symptoms.
Symptoms suggestive of heart failure include:
 Decreased exercise tolerance
Decreased exercise tolerance
 Dyspnea on exertion
Dyspnea on exertion
 Peripheral edema or ascites
Peripheral edema or ascites
 Orthopnea
Orthopnea
 Paroxysmal nocturnal dyspnea
Paroxysmal nocturnal dyspnea
 Unexplained confusion or altered mental status in an elderly patient (as a result of decreased cerebral perfusion)
Unexplained confusion or altered mental status in an elderly patient (as a result of decreased cerebral perfusion)
 Abdominal symptoms associated with ascites or hepatic engorgement (e.g., nausea, abdominal pain, or early satiety)
Abdominal symptoms associated with ascites or hepatic engorgement (e.g., nausea, abdominal pain, or early satiety)
If symptoms of progressive dyspnea, orthopnea, or paroxysmal nocturnal dyspnea are present in conjunction with a past cardiac history, the likelihood that the patient has heart failure dramatically increases. Patients with heart failure should be evaluated for activity tolerance, dyspnea, orthopnea, paroxysmal nocturnal dyspnea, rapid weight gain, palpitations, presyncope or syncope, and defibrillator shocks, as well as adherence to the medication regimen and dietary restrictions, at each encounter.
The physical examination can provide important information about the etiology of the patient’s symptoms and volume status to aid in the selection of appropriate therapies. Physical signs suggesting heart failure include:
 Elevated jugular venous pressure or positive hepatojugular reflux
Elevated jugular venous pressure or positive hepatojugular reflux
 Third heart sound (positive S3)
Third heart sound (positive S3)
 Laterally displaced apical impulse
Laterally displaced apical impulse
 Bibasilar pulmonary rales that do not clear up with cough (this finding is rare in patients with chronic heart failure)
Bibasilar pulmonary rales that do not clear up with cough (this finding is rare in patients with chronic heart failure)
 Peripheral edema not caused by venous insufficiency
Peripheral edema not caused by venous insufficiency
 Hepatomegaly and/or ascites
Hepatomegaly and/or ascites
In addition, weight, supine and upright blood pressure, heart rate, and temperature of extremities should be assessed at each encounter.
 DIAGNOSTIC STUDIES
DIAGNOSTIC STUDIES
Diagnostic studies for evaluating patients with suspected heart failure and for ongoing monitoring of the clinical status of patients with heart failure are listed in Table 10.5 (Yancy et al., 2013). Many of these tests are recommended to rule out other diseases that have clinical symptoms similar to those of heart failure and to delineate the underlying causes of heart failure so that they can be managed properly to reverse or prevent further progression of symptoms.
Central or obstructive sleep apnea is common in patients with heart failure. Patients with heart failure rarely report daytime somnolence. When a clinical suspicion exists, a patient should be referred for a sleep study. Treatment of obstructive sleep apnea with continuous positive airway pressure (CPAP) has demonstrated improvement in cardiac function and quality of life in patients with heart failure (Yancy et al., 2013).
Biomarkers
Biomarkers are useful in heart failure management. BNP and its amino acid N-terminal cleavage equivalent, NT-proBNP, are produced by cardiomyocytes in response to myocardial stretch. Assays for BNP and NT-proBNP are commonly used to aid in clinical decision making to diagnose or exclude heart failure. These biomarkers have a high negative predictive value. In the setting of dyspnea of unclear etiology, a BNP of <100 pg/mL or NT-proBNP of <300 pg/mL is associated with a decreased likelihood for heart failure as the cause of dyspnea. Patients with heart failure and dyspnea will likely have a BNP >400 pg/mL. NT-proBNP cutoff levels for diagnosing heart failure increase with age. For patients <50, 50 to 75, and >75 years of age, the optimal NT-proBNP cutoffs for diagnosing heart failure are 450 pg/mL, 900 pg/mL, and 1,800 pg/mL, respectively (Colucci & Chen, 2012). Although higher levels have a reasonable association with heart failure, other cardiac and noncardiac causes may result in high levels of these biomarkers. Heart failure treatment generally results in lower BNP or NT-proBNP levels over time. Other biomarkers, such as soluble ST2 and galectin-3, are being examined for their prognostic value in heart failure management; their use in routine clinical practice is still being evaluated.
 TREATMENT OPTIONS, EXPECTED OUTCOMES, AND COMPREHENSIVE MANAGEMENT
TREATMENT OPTIONS, EXPECTED OUTCOMES, AND COMPREHENSIVE MANAGEMENT
Heart failure management is driven by current clinical practice guidelines. In the United States, these guidelines are developed by two main task forces: the American College of Cardiology Foundation/American Heart Association (ACCF/AHA; circ.ahajournals.org/content/early/2013/06/03/CIR.0b013e31829e8807.citation) and the Heart Failure Society of America (www.heartfailureguideline.org/). A summary of these guidelines is presented in this chapter.
Heart Failure Classification
The ACCF/AHA and the New York Heart Association (NYHA) have developed classification systems for heart failure (Tables 10.6 and 10.7). Both systems provide useful and complementary information about the presence and severity of heart failure. The ACCF/AHA stages of heart failure emphasize the development and progression of disease and can be used to describe individuals and populations, whereas the NYHA classes focus on exercise capacity and symptomatic status of the disease (Yancy et al., 2013).
ACCF/AHA Stage A HFrEF
Primary prevention of heart failure should focus on prevention and treatment of underlying conditions associated with an increased risk of development of heart failure. Table 10.1 lists the etiologies for heart failure development. Ischemic heart disease/MI, hypertension, diabetes mellitus, and obesity encompass modifiable risk factors that may reduce the incidence or severity of heart failure. To understand how to prevent ischemic heart disease/MI and hypertension, one must first evaluate for risk factors for developing these conditions. To prevent heart failure, it is important to emphasize reduction in dietary fat and sodium consumption, weight maintenance, regular physical activity, and smoking cessation. In patients who already have a history of ischemic heart disease/MI, prevention of disease progression is also crucial (refer to Chapter 9, “Dyslipidemias”; Chapter 8, “Coronary Artery Disease”; Chapter 11, “Hypertension”; and Chapter 15, “Diabetes Mellitus,” for evidence-based guidelines related to the prevention, diagnosis, and management of these conditions).



