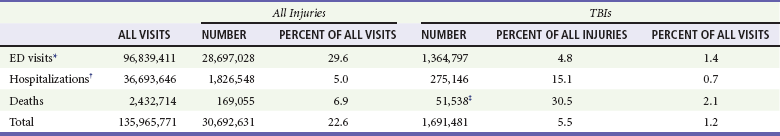
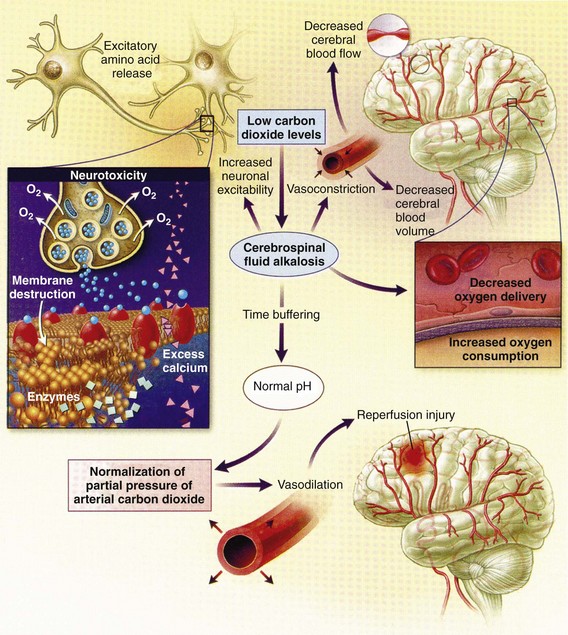
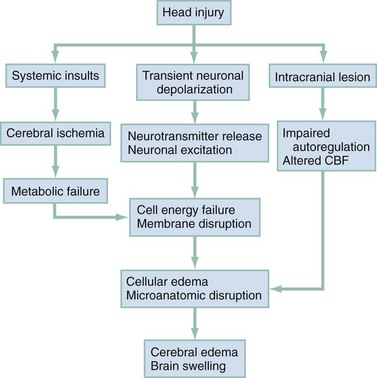
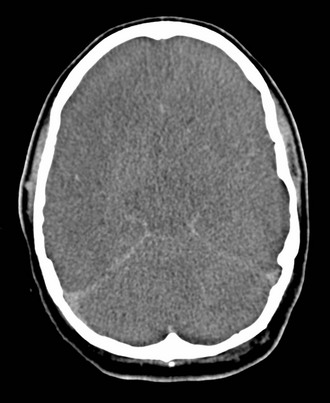
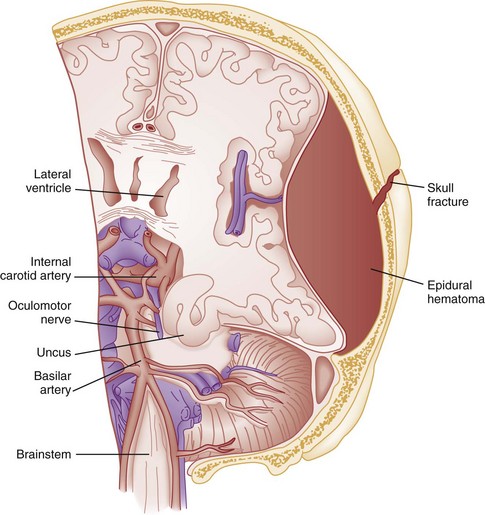
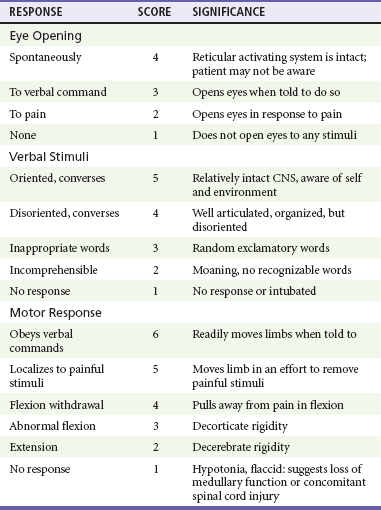
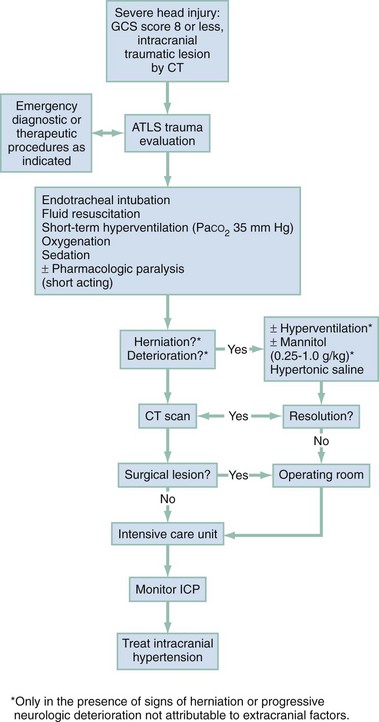
Chapter 41 The devastating consequences of head injury have been recorded since ancient times. Early neurosurgical records were primarily observational, with very few suggestions for treatment (Fig. 41-1). Despite centuries of investigation and the development of new and better intensive care, we still have not discovered effective therapies that can be applied after the injury to reverse most pathologic aspects of traumatic brain injury (TBI). In 1996, Congress approved the Traumatic Brain Injury Act (Public Law 104-166), which charged the Centers for Disease Control and Prevention (CDC) with “determining the incidence and prevalence of traumatic brain injury in all age groups in the general population of the United States.”1 Each year, an estimated 1.7 million people sustain head injury in the United States.1 Of these, 1.36 million (1.4% of all emergency department [ED] visits) undergo emergency evaluation, including approximately 470,000 children younger than age 14 years. TBI accounts for 15.1% of all injury-related hospitalizations in the United States. Overall, 80% sustain minor head trauma (Glasgow Coma Scale [GCS] score of 14 or 15), 10% have moderate head injuries (GCS score of 9-13), and 10% have severe head injuries (GCS score of 8). Almost 20% of all head-injured patients are hospitalized, and approximately 52,000 patients die each year from TBI (Table 41-1). Table 41-1 Traumatic Brain Injury (TBI) as a Portion of All Injuries and Emergency Department (ED) Visits *Persons who were hospitalized, died, or were transferred to another facility were excluded. †In-hospital deaths and patients who transferred from another hospital were excluded. ‡128 mortality records (from 2002-2006) were omitted because of missing age information. (Adapted from Faul M XL, Wald MM, Coronado VG: Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002-2006. Atlanta, Centers for Disease Control and Prevention, National Center for Injury Prevention and Control, 2010, pp 1-74.) The leading causes of head injury in the civilian population are falls (43.7%) and motor vehicle collisions (MVCs) (21.5%).1 TBI caused by blasts has been called the signature injury of the wars in Iraq and Afghanistan: TBI of any severity is estimated to affect as many as 10 to 20% of wartime service members.2–4 Head injury is the leading cause of traumatic death in patients younger than 25 years and accounts for nearly one third of all trauma deaths.1 Head injury from child abuse is common.5 New data suggest that with children younger than 12 months of age, TBIs and/or fractures attributable to abuse occurred in 1 of 2000 infants.6 The CDC estimates that there are at least 5.3 million Americans currently experiencing some degree of disability as a result of TBI. As veterans return to the United States, the number of patients experiencing the consequences of TBI has increased markedly. The skull comprises the frontal, ethmoid, sphenoid, and occipital bones and two parietal and two temporal bones. The unique layered architecture of the bones of the skull enhances its strength. Each bone consists of solid inner and outer layers separated by a layer of cancellous bone tissue (the diploë). In adults the bones of the skull average 2 to 6 mm in thickness; the bones in the temporal region are usually the thinnest of the skull.7 The cranial bones form a smooth outer surface of the skull, but within the cranial vault are many bone protrusions and ridges. Contrecoup injuries and contusions far from the site of head impact often occur as the accelerating brain strikes against these uneven bone surfaces. The cranial vault is rigid and nonexpandable, with an average volume in adults of approximately 1900 mL.8 Cranial contents exit or enter the skull through many foramina. The largest, the foramen magnum, is the site of exit of the brainstem and spinal cord from the cranium. Blood-Brain Barrier.: The blood-brain barrier (BBB) maintains the microenvironment of the brain tissue. Extracellular ion and neurotransmitter concentrations are regulated by movement across this barrier. When the BBB is intact, the ability of neuroactive drugs to penetrate into the brain tissue usually depends on their lipid solubility. Post-traumatic cerebral edema and possibly the biomechanics of the injury itself can cause a prolonged disruption of the BBB for up to several hours after trauma.9,10 Prolonged disruption of the BBB contributes to the development of post-traumatic vasogenic cerebral edema. The brain has an extremely high metabolic rate, using approximately 20% of the entire oxygen volume consumed by the body. To provide for its high metabolic demands, the brain requires approximately 15% of the total cardiac output. Optimal regional CBF is maintained by the ability of cerebral vessels to alter their diameter in response to changing physiologic conditions.10 Hypertension, alkalosis, and hypocarbia promote cerebral vasoconstriction; hypotension, acidosis, and hypercarbia cause cerebral vasodilation. In the normal brain, CBF is maintained at constant levels with a mean arterial pressure (MAP) of 60 to 150 mm Hg. This is referred to as autoregulation. Outside this range, the CBF varies linearly with MAP. Cerebral vasoactivity is very sensitive to changes in systemic carbon dioxide and oxygen partial pressures (PCO2 and PO2, respectively). The response to changes in PCO2 is nearly linear between PCO2 values of 20 and 60 mm Hg.11 In this range, lowering PCO2 by as little as 1 mm Hg decreases the diameter of cerebral vessels by 2 or 3%, which corresponds to an overall change in CBF of 1.1 mL per 100 g of tissue per minute. The physiologic response of blood vessels to PCO2 is the rationale for the acute use of brief hyperventilation to control increased intracranial pressure (ICP) after head injury. As PCO2 decreases with hyperventilation, cerebral vasoconstriction occurs. As a result, the volume of blood per unit area of brain tissue decreases. This decrease (even if small) may buffer the effects of increasing edema or an expanding hematoma within the rigid cranial vault. The vasoconstriction produced by extreme changes in PCO2 (20 mm Hg or less) can be so pronounced that some areas of brain experience ischemia; subsequently, tissue hypoxia can occur.10,12,13 Therefore hyperventilation is controlled and monitored, with a goal of maintaining the PCO2 between 30 and 35 mm Hg during prolonged hyperventilation, and is reserved for patients who are showing signs of acute herniation.12,13 Over 12 to 24 hours, injured vessels may lose their responsiveness to hyperventilation-induced hypocarbia and become vasodilated. Blood may then be shunted to the injured area, resulting in increased brain swelling and mass effect. Prolonged (i.e., beyond the acute resuscitation) or prophylactic hyperventilation is therefore not recommended as a treatment for increased ICP, and hyperventilation is not used for the routine management of head-injured patients with no signs of increased ICP.12,13 The neurologic effects of hypocapnia are illustrated in Figure 41-2. The cerebral vessels also respond to changes in PO2. As PO2 declines, cerebral vessels dilate to ensure adequate oxygen delivery to brain tissue. When brain injury has occurred, increased CBF in the presence of a disrupted BBB can promote the formation of vasogenic edema. Avoiding or reversing hypoxia is therefore an essential goal in the acute management of the head-injured patient.14 The responses of the cerebral vasculature to changing physiologic conditions protect the brain by increasing the delivery of oxygen to tissue, enhancing the removal of metabolic end products, and allowing nearly instantaneous adjustments of regional blood flow to meet the changing metabolic demands. Cerebral Perfusion Pressure.: CBF also depends on cerebral perfusion pressure (CPP), which is the pressure gradient across the brain. The determinants of CPP are MAP and the resistance to CBF produced by mean systemic venous pressure and ICP. Because ICP is higher than mean systemic venous pressure, ICP effects predominate. Therefore CPP is estimated as Direct Injury.: Direct impact head injury occurs when the head is struck by an object or its motion is arrested by another object. The resulting damage depends on the consistency, mass, surface area, and velocity of the object striking the head. Direct injury can also be caused by compression of the head. External signs of trauma are frequently noted at the site of application of the impact or compression force. The skull initially bends inward at the point of contact. If the force is sufficient, a skull fracture can occur. The cranium absorbs some of the applied energy, and some energy is transmitted to the brain by shock waves that travel distant to the site of impact or compression. These shock waves distort and disrupt intracranial contents and temporarily alter regional ICP as they propagate. In general, the more rapidly a force is applied, the greater the damage it causes. The extent of direct injury depends on the vasoelastic properties of the underlying region of brain tissue, the duration of the force applied, the magnitude of the force reaching the brain tissue, and the surface area of the brain that is affected by the application of the force. In cases of penetrating trauma, the mass, shape, direction, and speed of the penetrating object also affect the extent of direct injury. Indirect Injury.: In indirect brain injury, the cranial contents are set into motion by forces other than the direct contact of the skull with another object. A common example is acceleration-deceleration injury, such as the shaken impact syndrome.15,16 No direct mechanical impact is sustained, but the cranial contents are set into vigorous motion. The brain moves within the skull, and bridging subdural vessels are strained. Subdural hematomas (SDHs) may result. Differential acceleration of the cranial contents occurs, depending on the physical characteristics of the brain region. As one brain region slides past another, shear and strain injuries are produced. This movement results in diffuse injuries, such as diffuse axonal injury (DAI) or concussion. Additional injury occurs as the movement of the intracranial contents is abruptly arrested and the brain strikes the skull or a dural structure. Contrecoup contusions are an example of the injury produced in this manner. In penetrating injury the traversal of the object produces pressure waves that can strike structures distal to the path of the missile. Primary and Secondary Brain Injuries The circumstances and extent of the primary injury are not the only contributors to the final neurologic outcome after head injury. The traumatic event also produces injury at the functional and anatomic cellular level, which begins soon after the impact and continues for several hours and even days after injury. Secondary brain injury results from intracellular and extracellular derangements that are probably initiated at the time of trauma by a massive depolarization of brain cells and subsequent ionic shifts.17 Animal studies have revealed a complicated series of neurochemical, neuroanatomic, and neurophysiologic reactions after head injury (Fig. 41-3). The cell has some compensatory mechanisms to protect itself from widespread damage, such as endogenous free radical scavengers and antioxidants. With significant trauma, however, these systems are quickly overwhelmed, and the functional and structural integrity of the cell is threatened. Human studies document similar changes. The relative importance and contribution of each adverse reaction to the final functional status of the damaged cell are uncertain, as are the rate and duration of each detrimental event. All currently used acute therapies for TBI are directed at reversing or preventing secondary injury. Experimental evidence for many investigational agents aimed at specific steps in the destructive processes suggests that some aspects of secondary brain injury may be reversed or modified. Multiple ongoing head injury trials have been performed with numerous investigational therapeutic interventions; to date, none have proved useful in the clinical setting.18,19 The final neurologic outcome after head trauma is influenced by the extent and degree of secondary brain injury. In turn, the amount of secondary brain injury depends on certain premorbid and comorbid conditions, such as the age of the patient and trauma-related systemic events.19–21 A primary goal in the emergency care of the head-injured patient is prevention or reduction of systemic conditions that are known to worsen outcome after TBI. Common secondary systemic insults in trauma patients include hypotension, hypoxia, and anemia and hyperpyrexia. Hypotension, defined as a systolic blood pressure less than 90 mm Hg, has been found to have negative impact on severe head injury outcome.18,19,21,22 Systemic hypotension reduces cerebral perfusion, thereby potentiating ischemia and infarction. The presence of hypotension nearly doubles the mortality from head injury and worsens the outcome of the patients who survive.17,21–23 Hyperpyrexia (core body temperature of 38.5° C) is also correlated with worsened outcomes after TBI, and both its magnitude and its duration seem to contribute. The exact mechanism by which it causes damage is yet to be determined but likely involves stimulation of metabolism in injured areas of the brain, thus recruiting blood flow with a resultant increase in ICP.18,21 Hypoxia, defined as a PO2 less than 60 mm Hg, probably occurs often in the head-injured patient. Causes include (1) transient or prolonged apnea caused by brainstem compression or injury after the traumatic event; (2) partial airway obstruction caused by blood, vomitus, or other debris in the airway of the traumatized patient; (3) injury to the chest wall that interferes with normal respiratory excursion; (4) pulmonary injury that reduces effective oxygenation; and (5) ineffective airway management, such as the inability to bag-valve-mask or intubate the patient in an effective or timely manner. The exact incidence of hypoxia in the head-injured patient is difficult to estimate because it is often unnoticed or undocumented in the out-of-hospital setting. When its occurrence is documented, the overall mortality from severe head injury may double.18,22–24 Increased recognition of the potentially devastating consequences of hypoxia and associated hypercarbia has led to more vigilance in out-of-hospital and emergency settings. Anemia caused by blood loss can be detrimental to the head-injured patient by reducing the oxygen-carrying capacity of the blood, thus reducing the amount of necessary substrate delivered to the injured brain tissue. When anemia (hematocrit 30%) occurs in patients with severe head injury, the mortality rate increases.18,21 Other potential reversible causes of systemic insult in head injury include hypercarbia, coagulopathy, and seizures. Increased Intracranial Pressure ICP represents a balance of the pressures exerted by the contents of the cranial cavity. This relationship is explained by the Monro-Kellie doctrine.25,26 Because the craniospinal intradural space is almost nonexpandable, the sum of the volume of brain, CSF, and blood within the cranium will remain constant. If the volume of any of these components increases, the volume of another must decrease for a constant ICP to be maintained. Increased ICP is defined as CSF pressure greater than 15 mm Hg (or 195 mm H2O) and is a frequent consequence of severe head injury. Initially, as ICP increases as a result of a traumatic mass lesion or edema formation, the CSF is displaced from the cranial vault to the spinal canal, offsetting the increased blood or brain volume. When this compensatory mechanism is overwhelmed, the elastic properties of the brain substance allow tissue compression to provide buffering for the increasing pressure. Depending on the location and the rate of expansion of the traumatic mass lesion and the rate of cerebral edema formation, the intracranial compensatory mechanisms can accommodate an increased volume of 50 to 100 mL. Beyond that, even small additional changes in intracranial volume relationships, such as those caused by vasodilation, CSF obstruction, or small areas of focal edema, cause a dramatic increase in ICP. If ICP increases to the point at which CPP is compromised, vasoparalysis occurs and autoregulation is lost. The CBF then depends directly on the systemic MAP. With the loss of autoregulation, massive cerebral vasodilation occurs. Systemic pressure is transmitted to the capillaries, and the outpouring of fluids into the extravascular space can contribute to vasogenic edema and thus further increase ICP. If ICP rises to the level of the systemic arterial pressure, CBF ceases and brain death occurs. Methods to reduce elevated ICP include hyperventilation, use of osmotic and diuretic agents, and CSF drainage. Uncontrollable increased ICP is defined as an ICP of 20 mm Hg or higher refractory to treatment. If ICP is not controlled, herniation syndromes can occur, resulting in brainstem compression and subsequent cardiorespiratory arrest. In the United States, the use of ICP monitoring and control has become standard in cases of moderate and severe TBI despite the lack of prospective controlled research showing clear efficacy as an individual patient treatment modality.18 Two primary types of brain swelling occur after head injury: congestive brain swelling and cerebral edema. Congestive brain swelling results from an increased intracranial blood volume. Hyperemia occurs early after trauma and can persist for the first few days after injury.23,27 It is especially common in children. The increased blood volume is most likely caused by vasodilation, which occurs as a compensatory mechanism to maintain optimal CBF in the presence of increased metabolic needs of the damaged brain tissue. Cerebral edema is an increase in brain volume caused by an absolute increase in cerebral tissue water content. Diffuse cerebral edema may develop soon after head injury; however, its presence and extent do not always correlate with the severity of head injury. On computed tomography (CT) scans, diffuse edema manifests as bilateral compression of the ventricles, loss of definition of the cortical sulci, or effacement of the basal cisterns (Fig. 41-4). Focal edema adjacent to traumatic mass lesions demonstrates decreased density on CT scans compared with normal tissue. CT can also detect a mass effect, caused by edema surrounding a traumatic lesion. Both vasogenic and cytotoxic cerebral edema occur in the setting of trauma; the incidence and onset of each relative to the other depend on the nature of the injury. Vasogenic edema arises from transvascular leakage caused by mechanical failure of the tight endothelial junctions of the BBB.23,27 Vasogenic edema accumulates preferentially in white matter and can become widespread. It is frequently associated with focal contusions or hematomas. Vasogenic edema eventually resolves as edema fluid is reabsorbed into the vascular space or the ventricular system. Cytotoxic edema is an intracellular process that results from membrane pump failure. It is common after head injury and is frequently associated with post-traumatic ischemia and tissue hypoxia. Normal membrane pump activity depends on adequate CBF to ensure adequate substrate and oxygen delivery to brain tissue. If the CBF is reduced to 40% or less of baseline, cytotoxic edema begins to develop. If CBF drops to 25% of baseline, membrane pumps fail and cells begin to die. Congestive brain swelling can contribute to cytotoxic edema if it becomes severe enough to increase ICP and reduce CPP so that cerebral circulation cannot be maintained. Recent work suggests that cytotoxic cerebral edema is the predominant form of edema in patients who have experienced TBI.23,28 Progressive hypertension associated with bradycardia and diminished respiratory effort is a specific response to acute, potentially lethal increases in ICP. This response is called Cushing’s reflex or Cushing‘s phenomenon, and its occurrence indicates that the ICP has reached life-threatening levels. Cushing’s reflex can occur whenever ICP is increased, regardless of the cause. The full triad of hypertension, bradycardia, and respiratory irregularity is seen in only one third of cases of life-threatening increased ICP.23,29 Cerebral herniation occurs when increasing cranial volume and ICP overwhelm the natural compensatory capacities of the CNS (Fig. 41-5). Increased ICP may be the result of post-traumatic brain swelling, edema formation, traumatic mass lesion expansion, or any combination of the three. When increasing ICP cannot be controlled, the intracranial contents shift and herniate through the cranial foramen. Herniation can occur within minutes or up to days after TBI. When the signs of herniation syndrome are present, however, mortality approaches 100% without rapid implementation of temporizing emergency measures and definitive neurosurgical therapy. Uncal Herniation.: The most common clinically significant traumatic herniation syndrome is uncal herniation, a form of transtentorial herniation. Uncal herniation is often associated with traumatic extra-axial hematomas in the lateral middle fossa or the temporal lobe. The classic signs and symptoms are caused by compression of the ipsilateral uncus of the temporal lobe on the U-shaped edge of the tentorium cerebelli as the brain is forced through the tentorial hiatus. As compression of the uncus begins, the third cranial nerve (CN) is compressed; anisocoria, ptosis, impaired extraocular movements, and a sluggish pupillary light reflex develop on the side ipsilateral to the expanding mass lesion. This phase may last for minutes to hours, depending on how rapidly the expanding lesion is changing. As the herniation progresses, compression of the ipsilateral oculomotor nerve eventually causes ipsilateral pupillary dilation and nonreactivity. Initially in the uncal herniation process, motor examination findings can be normal, but contralateral Babinski’s responses develop early.23 Babinski’s sign is dorsiflexion of the great toe and fanning of the other toes. Contralateral hemiparesis develops as the ipsilateral peduncle is compressed against the tentorium. With continued progression of the herniation, bilateral decerebrate posturing eventually occurs; decorticate posturing is not always seen with the uncal herniation syndrome. Pupillary dilation and nonreactivity are the most reliable physical examination findings with regard to lesion location. Central Transtentorial Herniation.: The central transtentorial herniation syndrome is demonstrated by rostrocaudal neurologic deterioration caused by an expanding lesion at the vertex or the frontal or occipital pole of the brain. It is less common than uncal transtentorial herniation. Clinical deterioration occurs as bilateral central pressure is exerted on the brain from above. The initial clinical manifestation may be a subtle change in mental status or decreased level of consciousness, bilateral motor weakness, and pinpoint pupils (2 mm). Light reflexes are still present but are often difficult to detect. Muscle tone is increased bilaterally, and bilateral Babinski’s signs may be present. As central herniation progresses, both pupils become midpoint and lose light responsiveness. Respiratory patterns are affected, and sustained hyperventilation may occur. Motor tone increases. Decorticate posturing is elicited by noxious stimuli. This progresses to bilateral decorticate and then spontaneous decerebrate posturing. Respiratory patterns initially include yawns and sighs and progress to sustained tachypnea, followed by shallow slow and irregular breaths immediately before respiratory arrest. Cerebellotonsillar Herniation.: Cerebellotonsillar herniation occurs when the cerebellar tonsils herniate downward through the foramen magnum. This is usually caused by a cerebellar mass or a large central vertex mass causing the rapid displacement of the entire brainstem.23 Clinically, patients demonstrate sudden respiratory and cardiovascular collapse as the medulla is affected. Pinpoint pupils are noted. Flaccid quadriplegia is the most common motor presentation because of bilateral compression of the corticospinal tracts. Mortality from cerebellar herniation approaches 70%.23 Upward Transtentorial Herniation.: Upward transtentorial herniation occasionally occurs as a result of an expanding posterior fossa lesion. Level of consciousness declines rapidly. These patients may have pinpoint pupils from compression of the pons. Downward conjugate gaze is accompanied by the absence of vertical eye movements. The GCS is an objective method of following the patient’s neurologic status (Table 41-2). The GCS assesses a patient’s best eye, verbal, and motor responsiveness. It was developed for the clinical evaluation of head trauma patients at 6 hours after trauma, and all initial validation studies investigated its application at this time. It was designed for assessment of patients with isolated head trauma who were hemodynamically stable and adequately oxygenated.26 The GCS is only one aspect of the neurologic examination (e.g., the motor score reflects best limb movement, and it cannot detect subtle changes in mental status). However, because of its inter-rater reliability, reliance on objective clinical data, and ease of application, the GCS has become a standard acute measure of neurologic function in patients with altered mental status from any cause, including head trauma. The acute application (within 6 hours) of the GCS in head-injured patients has limitations. Hypoxia, hypotension, and intoxication can falsely lower the initial GCS score.26 Intubation lowers the patient’s GCS score by automatically assigning a score of 1 for verbal response, regardless of the actual contribution of head injury to the clinical examination. Periorbital edema from direct eye trauma may make assessment of spontaneous eye opening difficult. Extremity fractures or occult spinal cord injuries may interfere with the motor examination. Children and patients with difficulty communicating are challenging to assess with the GCS. The GCS may miss subtle mental status changes and does not assess brainstem reflexes or pupillary reflexes. Decisions regarding continued resuscitation of severely head-injured patients should not be based on the initial GCS score because of these limitations. Patients should be fully resuscitated, with evacuation of all surgical lesions, and remain hemodynamically stable, and not be intoxicated before the GCS can be used to predict their prognosis.26 The patient’s acute motor examination assesses for strength and symmetry. Paralysis obscures involuntary reflexes; attempts should be made to perform the motor examination before paralytic agents are given. Hemiparesis contralateral to a fixed and dilated pupil suggests herniation syndrome. A false-localizing motor examination can be caused by contralateral cerebral parenchymal injury occurring simultaneously with the expanding mass lesion or by Kernohan’s notch syndrome (compression of the contralateral cerebral peduncle). False-localizing motor signs can also be caused by extremity trauma, spinal cord, or nerve root injury that makes the examination painful or difficult. If the patient is not cooperative or is comatose, motor movement should be elicited by application of noxious stimuli. Any movement should be recorded. Voluntary purposeful movement must be distinguished from abnormal motor posturing. Decorticate posturing is abnormal flexion of the upper extremity and extension of the lower extremity. The arm, wrist, and elbow slowly flex, and the arm is adducted. The leg extends and internally rotates, with plantar flexion of the foot. Decorticate posturing implies injury above the midbrain. Decerebrate posturing is the result of a more caudal injury and therefore is associated with a worse prognosis.19 The arms extend abnormally and become adducted. The wrist and fingers are flexed, and the entire arm is internally rotated at the shoulder. The neck undergoes abnormal extension, and the teeth may become clenched. The leg is internally rotated and extended, and the feet and toes are plantar flexed. In the acute setting, brainstem activity is assessed by the patient’s respiratory pattern, pupillary size, and eye movements. The oculocephalic response (doll’s eyes maneuver) tests the integrity of the pontine gaze centers. This response cannot be tested until cervical spine fractures have been ruled out. The oculovestibular response (cold water calorics) also permits assessment of the brainstem. Comatose patients no longer demonstrate nystagmus when cold water is placed in the ear canal; the only response is tonic deviation of the eyes toward the instilled cold water.24 This response is dampened by cerumen or blood in the patient’s ear canal, and the tympanic membrane needs to be intact for this test to be performed. The head and neck should be carefully examined for external signs of trauma that may have also produced underlying TBI. A scalp laceration, contusion, abrasion, or avulsion may overlie a depressed skull fracture. Basilar skull fractures are usually diagnosed by the clinical examination (Box 41-1). Although not always related to severe brain injury, their presence implies that a significant impact force was sustained during head trauma. Carotid artery dissections caused by a hyperflexion-extension neck injury can occasionally be detected by auscultation of a carotid bruit.24 In these patients, a careful neurologic examination should assess for subtle asymmetry between the carotid arteries. The percentage of concurrent cervical spine injury in patients with severe head trauma may be as high as 10.2%.27 Often, other spinal regions are also injured. The neurosurgical literature defines severe head injury as TBI manifested by a postresuscitation GCS score of 8 or lower within 48 hours. In the emergency setting, however, this definition is not practical because the outcome for the patient beyond the initial resuscitation is not known. Most emergency medicine research defines severe head injury by a GCS score of 8 or lower at the acute presentation after injury. The presence of any intracranial contusion, hematoma, or laceration is also considered severe injury (see Fig. 41-5). Approximately 10% of all head-injured patients who reach the ED alive have severe head trauma.1,30 The clinical prognostic indicators in the acute setting are initial motor activity, pupillary responsiveness, the patient’s age and premorbid condition, and the occurrence of secondary systemic insult during the acute period.23,28,31 Up to 25% of these patients have lesions requiring neurosurgical evacuation.18 The prognosis cannot be reliably predicted by the initial GCS score or initial CT scan. The overall mortality in severe head trauma has improved over the years and now remains stable at approximately 35%.32,33 Mortality for children is lower. For nonsurvivors of head injury who reach the hospital alive, the average time to death is 2 days after trauma. Adult survivors of severe head trauma are usually severely disabled; currently, only 7% have moderate disability or a good outcome. Children older than 2 years who survive a severe closed head injury have a better outcome than adults. Head trauma can produce profound effects on the cardiovascular system if compression of the brainstem and medulla occurs. Any cardiac dysrhythmia can occur and produce cardiac instability.23,34 All head-injured patients should be placed on a cardiac monitor during transport from the accident scene. The secondary survey of the head-injured patient should include a search for external signs of head trauma. Scalp lacerations may bleed a large volume into a bulky dressing. A less bulky dressing should be used with firm constant manual pressure applied to avoid excessive blood loss. Many severely head-injured patients are initially combative or agitated. Transporting an agitated patient who is fighting against physical restraints may exacerbate physical injury, cause an increase in ICP, and interfere with appropriate stabilization and management. It may be necessary to use out-of-hospital sedation or neuromuscular blockade for control. The use of sedatives or neuromuscular blockade may influence the initial ED evaluation of the neurotrauma patient. Therefore the risks and benefits of this acute intervention are carefully considered and decisions made on a case-by-case basis. Out-of-hospital protocols allowing the use of sedative agents for selected agitated head-injured patients should be established. Currently used agents include lorazepam (Ativan), diazepam (Valium), midazolam (Versed), and certain butyrophenones (e.g., haloperidol, droperidol). Some investigators are looking at the use of ketamine for prehospital sedation in combative head-injured patients. Recent research questions whether the widely held belief that ketamine increases ICP and CBF is accurate.35 Currently, the existing prehospital data are insufficient to recommend ketamine use in combative prehospital head-injured patients. Severe head injury is the most common reason for helicopter transfers in trauma care. Although the decision to transport by helicopter should be made on a case-by-case basis, considerations for helicopter use from an accident scene include a long extrication time, ground transport of longer than 30 minutes to an appropriate ED and trauma care facility, two or more severely injured patients at a scene, and assistance in performing expedient lifesaving procedures, especially airway management. Several recent studies have supported that the use of helicopter emergency services improves outcomes in severely head-injured patients.24,36–38 Controversy exists regarding the benefits of out-of-hospital intubations in patients with severe and moderate head injuries. It is unclear whether field intubations truly improve neurologic outcome or survival. Unsuccessful attempts at field intubations may add to out-of-hospital time and increase the risk of aspiration or hypoxia.39–41 In 1997 Winchell and Hoyt showed that patients who had sustained severe head injuries and who were intubated in the out-of-hospital setting had an improved survival compared with those who were not intubated.29 Since that time, others have challenged this finding. In the San Diego paramedic rapid sequence intubation (RSI) trial, Davis and colleagues42 found an increase in mortality and morbidity in patients who sustained severe head injuries and underwent out-of-hospital RSI compared with matched historical controls.42 Potential explanations included frequent hypoxic episodes with associated bradycardia, unintentional hyperventilation, and prolonged scene times for those undergoing out-of-hospital RSI. Wang and colleagues found a fourfold increase in mortality among patients who sustained severe TBI and received ground ambulance intubation compared with ED intubation.24 Flight clinician out-of-hospital intubation has been associated with decreased mortality and improved neurologic outcome, likely because of the higher airway management training requirements of air medical programs.24,37–40 These studies are somewhat limited by lack of generalizability24,42–46 (e.g., most emergency medical service systems do not have RSI capabilities) or by the use of nonvalidated outcome measures of neurologic function.24 Out-of-hospital intubation carries the following risks: frequent hypoxic episodes during intubation attempts, with or without concurrent bradycardia; unintentional hyperventilation of intubated patients; prolonged scene times because of the time demands associated with the intubation process; and persistent hypotension, likely from other injuries that go unattended while the airway management occurs. However, hypoxia must be avoided in head-injured patients, and out-of-hospital airway protocols balance the risks of emergency intubation in an uncontrolled setting with the need to secure an airway at risk. The key to a successful out-of-hospital RSI program involves well-trained clinicians with specific RSI protocols, involved medical control, frequent continuing education, and consistent quality assurance and improvement. Fakhry and colleagues47 reported that their helicopter clinicians had a 96.6% RSI success rate with few complications and no esophageal intubations. Airway.: RSI is the preferred method for securing the airway in combative or agitated patients. If possible, a brief neurologic examination should be performed before the patient is given any sedative or neuromuscular blocking agents. In general, the agents used for RSI in the head-injured patient are the same as those used for other patients, although attention is given to the increased ICP that can potentially occur with any physical stimulation of the respiratory tract. Lidocaine (1.5-2 mg/kg intravenous push) may attenuate the cough reflex, hypertensive response, and increased ICP associated with intubation, although this is vigorously debated.47–49 If succinylcholine is used, premedication with a subparalytic dose of a nondepolarizing agent can be considered if time permits because fasciculations produced by succinylcholine may increase ICP. The degree of ICP elevation and its clinical significance are unclear, however, and are balanced against the need for rapid establishment of an airway. Etomidate (0.3 mg/kg intravenously [IV]), a short-acting sedative-hypnotic agent, has beneficial effects on ICP by reducing CBF and metabolism.50 In addition, etomidate has minimal adverse effects on blood pressure and cardiac output and fewer respiratory depressant effects than other agents. Concern has been raised that single-dose etomidate used during RSI may adversely affect patient outcome by modulating the inflammatory response and transient adrenal suppression.51 However, research both supporting and refuting this position exists, and no definitive answer has yet been established.51–56 Hypotension.: Hypotension is rarely caused by head injury except as a terminal event. If hypotension is detected at any time in the emergent management of a head-injured patient, a cause other than the head injury should be sought. Some important exceptions occur. Profound blood loss from scalp lacerations can cause hypovolemic hypotension. In infants, hemorrhage into an epidural or subgaleal hematoma can produce profound hypovolemic shock. In the presence of a concomitant high spinal cord injury, neurogenic hypotension may occur. This is rare, and usually the cord injury is apparent on physical examination. In less obvious cases, neurogenic hypotension can be differentiated from hypovolemic hypotension by its nonresponsiveness to fluid administration and by the presence of inappropriate bradycardia in the face of hypotension. Systemic hypotension cannot be tolerated in the head-injured patient without profound worsening of neurologic outcome; fluids or blood transfusion should therefore be delivered to maintain a systolic blood pressure of at least 90 mm Hg.22 The delivery of large amounts of fluid to severely head-injured patients who are hypotensive from other injuries does not produce clinically significant increases in ICP; fluids should never be withheld in the head trauma patient with hypovolemic hypotension for fear of increasing cerebral edema and ICP. The resuscitation goal is to keep the CPP at 50 or above. Hypotension may interfere with the accurate neurologic assessment of the brain-injured patient. Often, when blood pressure is restored, an improved neurologic status is observed. Traditionally, normal saline or lactated Ringer’s solution has been used for resuscitation of trauma patients with hypovolemic hypotension. Some research suggests that fluid resuscitation with hypertonic saline (HTS) rather than normal saline may improve neurologic outcome after TBI. However, recent data from the Resuscitation Outcomes Consortium showed no improvement in neurologic outcome when HTS was administered in the prehospital setting in severely head-injured patients not in hemorrhagic shock.57 As many as 50% of patients with severe head injury are victims of multiple trauma.22 The dramatic presentation of the head injury should not distract the clinician from a thorough search for other life threats. Hyperventilation.: Acute hyperventilation is a lifesaving intervention that can prevent or delay herniation in the patient with severe TBI while more definitive resources are mobilized. The goal is to reduce the PCO2 to the range of 30 to 35 mm Hg. Hyperventilation will reduce ICP by causing cerebral vasoconstriction; the onset of effect is within 30 seconds23 and probably peaks within 8 minutes after the PCO2 drops to the desired range. In most patients, hyperventilation lowers the ICP by 25%; if the patient does not rapidly respond, the prognosis for survival is generally poor. Prolonged hyperventilation is not recommended because it may cause profound vasoconstriction and ischemia. This vasoconstriction worsens CBF that is already severely compromised during the first 24 hours after TBI.12 Hyperventilation should be viewed as a short-term lifesaving intervention and should be used only when a patient experiences an acute neurologic decline or demonstrates signs consistent with herniation. Osmotic Agents.: Additional therapy for increased ICP includes the use of osmotic diuretics, such as mannitol and HTS. With deepening coma, pupil inequality, or other deterioration of the neurologic examination, osmotic agents may be lifesaving. Mannitol is the mainstay for control of elevated ICP in acute severe TBI. The Brain Trauma Foundation and the European Brain Injury Consortium recommend mannitol as the osmotic drug of choice.40,41 However, few comparative data exist on mannitol and other ICP-lowering medications.58 A Cochrane database review concluded that mannitol may have a small beneficial effect compared with pentobarbital.59 ICP-directed therapy based on neurologic signs may also be beneficial. However, other evidence suggests that mannitol may be detrimental compared with HTS.59,60 Further research is needed on optimal osmotic therapy in severe head trauma. Mannitol (0.25-1 g/kg) can effectively reduce cerebral edema by producing an osmotic gradient that prevents the movement of water from the vascular space into the cells during membrane pump failure and draws tissue water into the vascular space. This reduces brain volume and provides increased space for an expanding hematoma or brain swelling. The osmotic effects of mannitol occur within minutes and peak approximately 60 minutes after bolus administration. The ICP-lowering effects of a single bolus may last for 6 to 8 hours.61 Mannitol has many other neuroprotective properties. It is an effective volume expander in the presence of hypovolemic hypotension and therefore may maintain systemic blood pressure required for adequate cerebral perfusion. It also promotes CBF by reducing blood viscosity and microcirculatory resistance. It is an effective free radical scavenger, reducing the concentration of oxygen free radicals that may promote cell membrane lipid peroxidation. However, mannitol can produce renal failure or hypotension if given in large doses. It may also induce a paradoxical effect of increased bleeding into a traumatic lesion by decompressing the tamponade effect of a hematoma. Because of these and other potential problems, the use of mannitol should be reserved for head-injured patients with evidence of increasing ICP and neurologic deterioration.61 HTS has been used for severe TBI since 1919.62 Numerous clinical studies have demonstrated that HTS can significantly reduce ICP; however, the total number of enrolled patients in these trials is small.58,59,62,63 The interpretation of these clinical studies is complicated by variation in protocols, HTS concentration, and administration. Potential adverse events associated with HTS include renal failure, central pontine myelinolysis, and rebound ICP elevation.64 Encouraging clinical data are available on hospitalized pediatric TBI patients treated with a continuous infusion of 3% normal saline for control of intracranial hypertension.65,66 The clinical studies using HTS for acute field resuscitation of severe TBI have had conflicting results. Using a post hoc analysis of adult trauma data, Vassar and colleagues67 and Wade and colleagues68 showed beneficial effects of HTS on patients with severe TBI. However, Cooper and colleagues69 found no benefit for out-of-hospital use of HTS in reducing elevated ICP and improving CPP compared with lactated Ringer’s in hypotensive patients who had a head injury. As noted earlier, a large, multicenter, double-blinded, randomized, placebo-controlled clinical trial showed no improvement in neurologic outcome when HTS was administered in the prehospital setting in severely head-injured patients not in hemorrhagic shock.57 Barbiturates.: Barbiturate therapy is occasionally used in severely head-injured patients to reduce cerebral metabolic demands of the injured brain tissue. Barbiturates also affect vascular tone and inhibit free radical–mediated cell membrane lipid peroxidation. The effects of barbiturates are delayed relative to other acute interventions for reducing ICP; therefore they are rarely initiated in the ED. If other methods of reducing ICP have been unsuccessful, barbiturates may be added in the hemodynamically stable patient. Pentobarbital is the barbiturate most often used.70 Steroids.: Despite their popularity in the past, there is no benefit to giving steroids in head-injured patients. They do not lower ICP, and high-dose methylprednisolone in moderate and severe TBI is associated with increased mortality.71 Hypothermia.: Although hypothermia remains a significant area of research and promise for patients with severe and moderate TBI, the available scientific evidence is inconclusive with regard to improved mortality or morbidity with prophylactic hypothermia in patients with severe and moderate TBI. In a meta-analysis performed by the Brain Trauma Foundation, duration of hypothermic treatment for more than 48 hours was associated with a reduction in mortality. This finding is significantly limited owing to small sample sizes.72 A large trial of hypothermia use in moderate and severe TBI has recently begun in Europe. The Eurotherm3235 Trial will enroll 1800 patients over 41 months.73 Cranial Decompression.: Patients with signs of herniation who have not responded to other means of ICP reduction and who are rapidly deteriorating in the ED should be considered for placement of emergency burr holes. Emergency trephination has been described for centuries. It is a blind invasive procedure, and the chances of localizing the expanding lesions are uncertain. In patients who do not respond to hyperventilation and osmotic agents and have a unilateral dilated pupil with a history of “talk and deteriorate” after head trauma, emergency cranial decompression may temporarily reverse or arrest the herniation syndrome.74–77 In this setting, emergency trephination may allow enough time for a patient to undergo a formal craniotomy in the operating room. There has been renewed interest among neurosurgeons in the role of emergent decompressive craniectomy. Numerous papers have been published promoting more liberal use of decompressive craniectomy in patients with severe brain swelling and high ICPs.21 Outcome measurements have not been consistent, and results have been mixed. If performed, the procedure should be done early in the patient’s course. Two ongoing randomized studies may provide better direction regarding when this treatment is best indicated.21 Seizure Prophylaxis.: Up to 12% of all patients who sustain blunt head trauma and 50% of those with penetrating head injury develop early post-traumatic seizures (PTSs).78 Although the occurrence of seizures in the immediate post-trauma period has no predictive value for future epilepsy, early seizures can cause hypoxia, hypercarbia, release of excitatory neurotransmitters, and increased ICP, which can worsen secondary brain injury. Constantly firing neurons are soon depleted of their energy sources, and in the head trauma patient with compromised cerebral metabolism, uncontrolled seizures exacerbate the neurologic deficit.23 Box 41-2 lists accepted indications for early anticonvulsant therapy after head trauma. If the patient is actively seizing, benzodiazepines are administered as effective, rapid-acting first-line anticonvulsants. Lorazepam (0.05-0.1 mg/kg IV over 2-5 minutes up to a total of 4 mg) has been found to be most effective at aborting status epilepticus.23 Diazepam (0.1 mg/kg, up to 5 mg IV, every 10 minutes up to a total of 20 mg) is an alternative. For long-term anticonvulsant activity, phenytoin (18-20 mg/kg IV) or fosphenytoin (15-18 phenytoin equivalents/kg) can be given. Fosphenytoin has the advantages of rapid administration, a smaller volume of fluid for the dose delivered, and less hypotension than phenytoin. In a Cochrane review, the use of antiepileptic drugs reduced the risk of early seizures by 66%.79 Early seizure prophylaxis does not prevent late PTSs; the goal is to prevent additional insult to the damaged brain.78–80 Antibiotic Prophylaxis.: Infection may occur as a complication of penetrating head injury, open skull fractures, and complicated scalp lacerations. Prophylactic antibiotics may be used in these circumstances but are not recommended in patients with only otorrhea or rhinorrhea from a basilar skull fracture.81 Recombinant factor VIIa (rFVIIa) is a hemostatic agent that was originally developed to treat bleeding in hemophiliacs. Considerable interest has arisen regarding its potential use in intracerebral hemorrhage.82,83 rFVIIa costs approximately $1000/mg. Experience from the Iraq war and civilian use in multitrauma patients has produced conflicting results regarding its benefits in traumatic intracranial hemorrhage.82–85 Use of rFVIIa for traumatic head bleeds should be individualized and undertaken in concert with an institution’s treatment protocol. A recent review concluded that although off-label use of rFVIIa increased 140-fold from 2000 to 2008, no evidence exists that rFVIIa improves outcomes in five off-label indications. Specifically, the data regarding the use of rFVIIa in TBI are too limited for determination of the risks and benefits.86 Head-injured patients taking anticoagulants who require an emergent craniotomy may benefit from rFVIIa administration because it decreases the time to operating room intervention.82,83,85,86 Laboratory Tests.: The acute management of the severely head-injured patient is directed by physical examination and diagnostic imaging. Ancillary laboratory tests that may provide useful information in the subsequent management of the patient include a urine toxicology screen, blood alcohol level, complete blood count, electrolytes, glucose, and coagulation studies. Neuroimaging.: In the acute phase the most useful imaging technique is a non–contrast-enhanced head CT scan. This scan delineates acute intra-axial and extra-axial bleeding, subarachnoid blood, cerebral swelling, ischemic infarction caused by hypoxia after trauma, evidence of increased ICP, and pneumocephalus. Emergency management decisions are strongly influenced by these acute CT scan findings. The bone windows of the CT scan can detect skull fractures (including basilar fractures); plain skull radiographs are not necessary in patients who undergo CT scanning. Consultation.: All patients with severe head trauma require an imaging modality to determine the extent and nature of the brain injury and the necessity of neurosurgical intervention. Neurosurgical consultation should be obtained as soon as possible to help direct the patient’s subsequent management. Transfer.: Severely head-injured patients require admission to an institution capable of intensive neurosurgical care and acute neurosurgical intervention. If this is not available at the receiving hospital, the patient should be transferred to an appropriate institution by the most expedient transport method available.
Head Injury
Perspective
Principles of Disease
Scalp and Cranium
Brain and Cerebrospinal Fluid
Brain Cellular Damage and Death
Secondary Systemic Insults
Pathophysiology
Brain Swelling and Cerebral Edema
Cushing’s Reflex
Clinical Features and Diagnostic Strategies
Acute Neurologic Examination
Glasgow Coma Scale
Motor Examination: Posturing
Brainstem Function
Other Examination Findings
Management
Out-of-Hospital Care
Emergency Department (Fig. 41-6)

Full access? Get Clinical Tree


Head Injury

Only gold members can continue reading. Log In or Register to continue




