Chapter 91 Haemostatic failure
Failure of haemostasis is common in critically ill patients and may be complex and multifactorial in pathogenesis. As haemostatic failure may complicate a wide range of medical, surgical and obstetric disorders, definitive diagnosis and specific therapy can significantly impact on outcome. Frequently, complex tests are required for definitive diagnosis, but the urgency of the situation cannot always wait for the results, and therapy may be initiated on clinical evidence with minimal laboratory information. Consultation with a clinical haematologist is strongly recommended.
NORMAL HAEMOSTASIS
The haemostatic system is a delicately controlled component of the host defence system, the role of which is to initiate haemostasis where and when required and in adequate, but not in excessive, amounts. The system closely interacts with other components of the host defence system, including the acute stress responses, inflammation, healing and immune functions. There has been a paradigm shift in our understanding of the haemostatic system from one of interacting humoral factors to an integrated cell-based system.1,2 In view of the increasing range of focused therapies available that act on the haemostatic system to modulate its activity, it is important that core aspects of the structure and function of the system are summarised.
The conversion of blood from its fluid to solid state is an increasingly understood physiological process. The triad of vascular constriction, platelet plugging and fibrin clot formation forms haemostatic plugs and provides the framework on which haemostasis operates (Figure 91.1) and sets the scene for healing to occur. Thrombin is the potent proteolytic enzyme of the coagulation sequence converting fibrinogen to fibrin-soluble monomers, which subsequently polymerise to form the fibrin clot. Fibrinogen is the bulk protein of the coagulation system and fibrin is the end-product of this cascade of proteolytic activity, in which precursor coagulation proteins are activated to potent proteolytic enzymes, with the aid of cofactors, to produce active precursors further down the coagulation ‘amplifier’. The polymerised fibrin is further acted on by factor XIII to form a stable fibrin clot. The process of thrombin generation in small amounts initially occurs in relationship to tissue factor-bearing cells and the process is subsequently transferred to the activated platelet surface where amplification occurs.
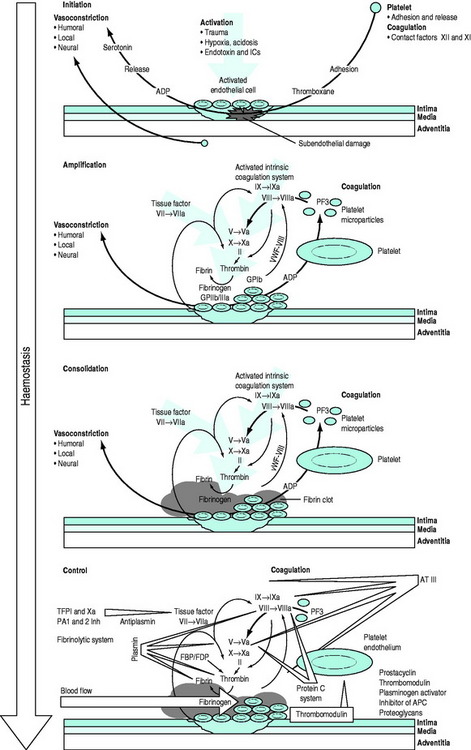
Figure 91.1 Overview of the haemostatic system. APC, activated protein C; AT III, antithrombin III; FBP, fibrinogen breakdown product; FDP, fibrinogen degradation product; ICs, immune complexes; TFPI, tissue factor pathway inhibitor; vWF, von Willebrand factor.
Following injury, vascular constriction reduces bleeding, and allows time to initiate haemostasis. With large volume haemorrhage, resulting systemic hypotension is an important physiological mechanism to minimise blood loss and facilitate stabilisation of the haemostatic plug. Controlled or ‘tolerated’ hypotension is now accepted as an important aspect of managing critical haemorrhage.3,4 This vascular constriction is further accentuated by vasoconstrictors released in association with platelet plug formation. Vascular endothelial cells play an active part by synthesising substances which act at the membrane surface and/or interact with platelets and the coagulation system (e.g. prostacyclin, antithrombin III, plasminogen activator, von Willebrand factor, thrombomodulin, heparin cofactor II and nitric oxide).5 Following the initial vascular reactions, successful haemostasis depends on adequate numbers of functioning platelets, coagulation cascade function, and poorly understood contributions from red cells and leukocytes.
Von Willebrand factor (vWF) is a multimeric glycoprotein that plays a central role in haemostasis by mediating adhesion of platelets to the exposed subendothelium and linking the primary vascular/platelet phase with coagulation by being the carrier protein for coagulant factor VIII which dissociates from vWF to form a complex on the activated platelet surface with IXa (tenase complex) to activate factor X to Xa. Further vWF is released from Weibel–Palade bodies in nearby endothelial cells and platelet α-granules. Platelets interact with vWF via their surface glycoprotein Ib-IX-V complexes. This slows their motion along the subendothelium, allowing receptor interaction leading to platelet binding to collagen. Adhesion to collagen is facilitated by the GPIa-IIa receptor. GPIIb-IIIa can then bind vWF, fibrinogen, fibronectin, vitronectin and thrombospondin, providing further important sites of anchorage for the spreading platelet. The initiation phase of platelet plug formation leads to the development of a platelet monolayer over the injured subendothelium. With the concomitant activation of the coagulation cascade there is local generation of thrombin. Thrombin is a potent platelet agonist via the protease-activated receptor (PAR) to produce a haemostatic plug. Further extension occurs by platelet recruitment by platelet agonists, such as thrombin and mediators, adenosine 5-diphosphate (ADP) and thromboxane A2 released directly from platelets. P-selectin is expressed during activation and also plays a role in platelet-to-platelet cohesion.
Parallel to and within the coagulation system are complex feedback mechanisms to ensure fine-tuning and protection against inappropriate and excessive activation.6–8 There are several inhibitory proteins, including antithrombin III, thrombomodulin, proteins C and S, tissue factor pathway inhibitor as well as the fibrinolytic system, which are all important in controlling the degree and site of fibrin formation. Indeed, thrombin itself acts as either a procoagulant or an anticoagulant depending on context. Massive activation, as may be seen in trauma and severe infections, can precipitate systemic activation, but in general, the system is well controlled and activity is localised to the area of the stimulus. Perturbations in this complex defence system can produce a wide range of clinical disorders from excessive arterial or venous thrombosis, microvascular obstruction and atheroma to haemostatic failure.
SYSTEMIC HAEMOSTATIC ASSESSMENT
There may be clinical features suggesting local or generalised failure of the haemostatic system (Figure 91.2). Clinical history is important, especially with respect to previous bleeding problems, family history, comorbid medical conditions and medications. The nature of surgery or an invasive intervention may have haemostatic issues that need specific consideration.
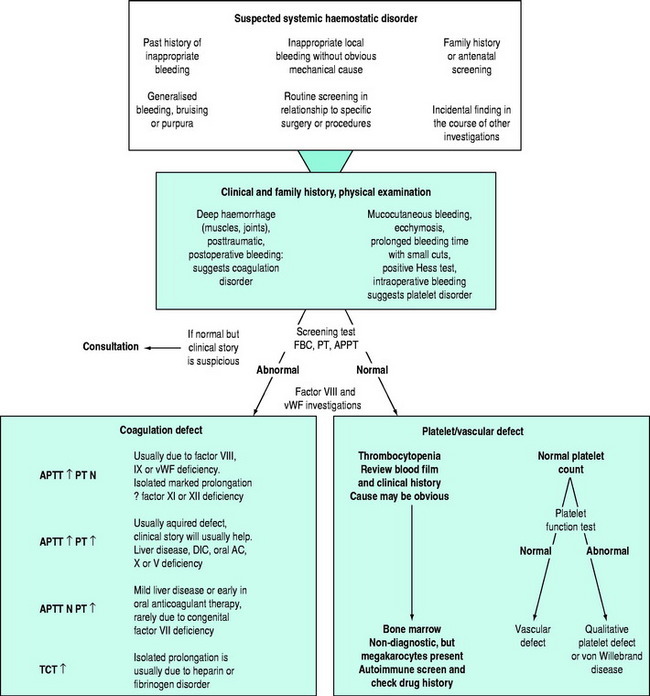
TESTS OF THE HAEMOSTATIC SYSTEM
Tests of whole blood clotting time and clot observation do not generally have a role in assessing haemostasis. However, in emergency settings, while waiting for laboratory results, observation of blood collected into a glass tube and maintained at 37°C for clot formation, size, retraction and possible lysis may provide crude information of a possible coagulopathy. The thromboelastogram is a more accurate and controlled procedure for ‘holistic’ assessment of the haemostatic system at the bedside, but requires close attention to technique and quality control. In most clinical settings, laboratory haemostatic screening tests are readily available, with near-patient testing techniques continuing to improve. A full blood count, prothrombin time (PT), activated partial thromboplastin time (APTT), fibrinogen level, D-Dimer ± thrombin clotting time (TCT) provide a broad screen for most clinically significant haemostatic disorders9–11 (Figure 91.3). Based on these results, and following consultation with a haematologist, further specific tests of haemostasis may be performed (e.g. mixing studies, factor assays, platelet function tests and test of fibrinolytic function). In broad terms:
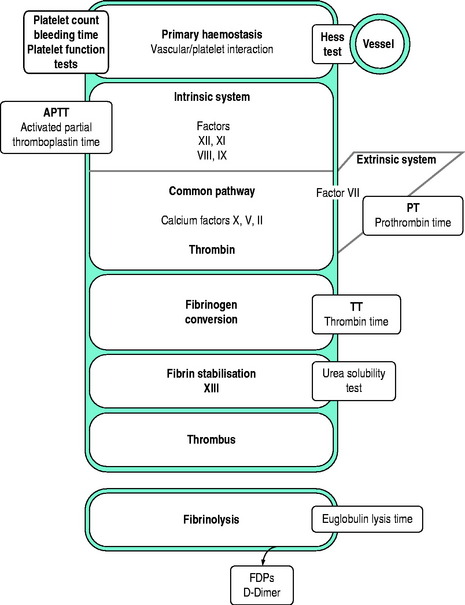
Figure 91.3 Investigations for a suspected haemostatic defect. FDPs, fibrinogen degradation products.
There are several important principles in the collection of blood samples for haemostatic investigations. Most samples are collected into citrated tubes and the amount of anticoagulant in the tube is related to the intended amount of blood to be collected into the tube. It is important that the correct amount of blood is added to the tube and gently mixed. Attention to venepuncture technique is crucial and rapid transfer of the sample to the laboratory is important. Contamination of the sample with tissue factor due to traumatic venepuncture will activate the sample and invalidate results. Collection from vascular access lines may result in diluted or heparin-contaminated samples. Incorrect laboratory results may be more dangerous to a patient than no results at all.
BLEEDING TIME (BT) (NORMAL 3–9 MINUTES)
The bleeding time is progressively prolonged when the platelet count falls below 75–100 × 109/l assuming platelet function is normal. It is also prolonged when platelet function is defective, in capillary/vascular disease and in some coagulation defects. The bleeding time is an invasive and operator-dependent test. It is not good for screening unselected patients and has generally been replaced by the platelet function analyser, PFA-100(R).
D-DIMER (NORMAL < 0.25 ng/l)
The D-Dimer assay measures cleavage fragments resulting from the proteolytic action of plasmin of fibrin. Elevation of D-Dimer may be seen in the postoperative state, trauma, renal impairment, sepsis and venous thrombosis. High levels of D-Dimer suggest excessive fibrinolysis, as seen in disseminated intravascular coagulation. The test is specific for fibrinolysis and not primary fibrinogenolysis.
PLATELET FUNCTION TESTS
Platelet function tests are used for the diagnosis of platelet disorders.12,13 In recent years there has been increasing interest in the use of platelet function tests to monitor antiplatelet therapy. Unfortunately, platelet function testing is poorly standardised and good quality control is commonly lacking. The diagnostic utility of some of the newer simpler assays of platelet function have been evaluated in a range of clinical settings, but their value remains controversial.
A popular technology is the platelet function analyser (PFA-100) in which citrated whole blood is placed in a disposable cartridge containing a membrane coated with collagen/adrenaline or collagen/ADP into which a microscopic aperture has been cut. With high shear rates contact of blood with the membrane causes platelets to aggregate and occlude the aperture. The time to occlude the aperture is recorded as the end-point and referred to as the closure time.14 The technique is simple and rapid, but subject to a number of variables such as the platelet count, haematocrit, ABO blood group and plasma vWF levels. A normal result has a high negative predictive value and is increasingly becoming popular as a screening test for platelet dysfunction; however, false positive and negative results may occur. The test is more sensitive than the bleeding time which is now generally regarded as an obsolete test.
THROMBOELASTOGRAM (TEG)
Thrombelastography was originally described 50 years ago, but failed to establish a role in routine laboratory haemostasis testing. Improvements in the technology, especially standardisation and computerisation, have now made the TEG a valuable tool for monitoring haemostasis as a holistic dynamic process supplementing the long-standing conventional coagulation screens. The TEG measures the viscoelastic properties of blood as it clots under low shear conditions. The patterns of changes in shear elasticity provide information about the kinetics of clot formation, growth, strength and stability of the formed clot. The TEG is increasing being used as a point of care test, providing ‘real time’ information about a patient’s haemostatic function.15
CONGENITAL HAEMOSTATIC DEFECTS
THE HAEMOPHILIAS
HAEMOPHILIA A (CLASSIC HAEMOPHILIA)
Haemophilia A is an X-linked disorder due to a deficiency of factor VIII. The mainstay of haemophilia treatment is coagulation factor replacement.16,17 Coagulation factor concentrates or recombinant factor VIII are given to prevent bleeding and to control existing haemorrhage. The dose of clotting factor is calculated on the basis that one unit of coagulation factor is that amount present in 1 ml of pooled normal plasma. An individual with a plasma volume of 3000 ml would have 3000 U of the clotting factor in the circulation. If such person was a haemophiliac with <1% factor VIII level in the plasma, a dose of 3000 U of factor VIII would be required to raise the plasma level to 100% of normal.
VON WILLEBRAND’S DISEASE
Classification of vWD is complex and controversial. Therapy may be determined by the specific subtype and expert haematological consultation is important.18,19 Clinical heterogeneity is reflected in the classification of subtypes corresponding to specific pathophysiological mechanisms.
CONGENITAL PLATELET DISORDERS
Congenital platelet disorders are rare, the commonest severe disorder being Glanzmann’s thrombaesthenia.20 Platelet transfusions may be required for acute bleeds or in relation to elective surgery; DDAVP ± antifibrinolytic therapy and recombinant factor VIIa may all have a role.
ACQUIRED HAEMOSTATIC DISORDERS
CRITICAL HAEMORRHAGE AND MASSIVE BLOOD TRANSFUSION
The nature and management of haemostatic defects secondary to acute and/or massive blood loss and transfusion remains poorly understood.21 The coagulopathy of trauma is characterised by non-surgical bleeding from mucosal lesions, serosal surfaces, wounds and vascular access sites. Hypothermia, acidosis and haemodilution are commonly present, occasionally with features of disseminated intravascular coagulation (DIC). DIC typically occurs acutely after trauma when brain, fat, amniotic fluid or other potent thromboplastins enter the circulation. It may occur less acutely when endothelial inflammation or failure reduces clearing of activated coagulation factors. The coagulopathy of trauma should be anticipated in massive transfusion situations and there is increasing support for early treatment with plasma, which may prevent or delay its onset.
Further aggravation of the complications of massive blood transfusion can be avoided or minimised if correctable defects in the haemostatic system are identified (Table 91.1). The relative importance of the different potential mechanisms responsible for haemostatic failure is difficult to determine, but the identification of correctable defects will avoid or minimise complications. Haemostatic failure correlates poorly with the volume of transfusion or components administered, but better with the nature of the insult, degree of hypovolaemia, and time to resuscitation. Trauma patients with major coagulopathy and microvascular haemorrhage usually have abnormal laboratory parameters prior to massive blood transfusion. Except for severe abnormalities, haemostatic laboratory parameters correlate poorly with clinical evidence of haemostatic failure. Thrombocytopenia and impaired platelet function are probably the most consistent significant haematological abnormalities, correction of which may be associated with control of microvascular bleeding.
Table 91.1 Possible factors contributing to haemostatic failure following massive blood transfusion
< div class='tao-gold-member'> Only gold members can continue reading. Log In or Register to continue
Related posts:
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|

