KEY POINTS
The physiological regulation of blood glucose involves hormonal and neural mechanisms, resulting in a control of glucose flux across cell membranes.
High glucose concentrations are associated with cellular toxicity.
Stress hyperglycemia is a marker of severity of illness.
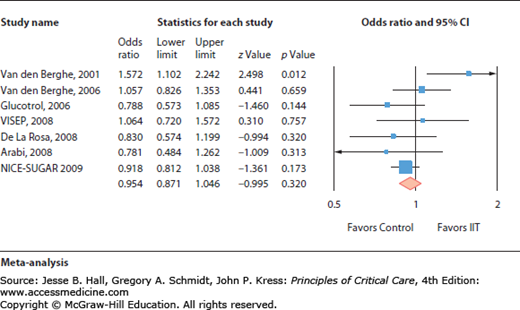
Intensive insulin therapy used to tightly control blood sugar was found beneficial in one single-center study, but not in seven independent other prospective trials.
Current recommendations advocate a moderate glucose control by insulin therapy.
INTRODUCTION
Critical illness is typically associated with a so-called stress-induced hyperglycemia, defined as a transient hyperglycemia during illness in patients without previous evidence of diabetes.1 The relationship between stress hyperglycemia and poor outcome is largely established. This association reflects the validity of blood glucose concentration as a marker of illness severity. However, the correction of a moderate stress hyperglycemia may improve the prognosis. Indeed, in 2001, a large randomized controlled trial in critically ill surgical patients demonstrated that tight glucose control (TGC) (defined as the restoration and maintenance of blood glucose between 80 and 110 mg/dL) by intensive insulin therapy (IIT) was associated with a decreased mortality and rate of complications.2 However, subsequent studies performed in other intensive care units (ICU)3-8 failed to reproduce the beneficial effects of IIT titrated to achieve TGC.
These conflicting results raise the clinically relevant question: How to control glycemia in ICUs? This chapter intends to summarize the current understanding of the physiological regulation of glycemia, the toxicity of hyperglycemia, the mechanisms and consequences of stress hyperglycemia, and the available clinical data from observational and interventional studies and to discuss the unsolved issues and the implications for the daily clinical practice. Updated formal recommendations will be suggested for glucose control in critically ill and postoperative patients.
PHYSIOLOGICAL REGULATION OF BLOOD GLUCOSE
Blood glucose concentration (BG) is tightly regulated by two types of mechanisms1:
The hormonal system consists in a balance between insulin, which will induce the entrance and utilization of glucose into tissues, and the so-called “hyperglycemic” counterregulatory hormones (glucagon, epinephrine, cortisol).
The neural mechanism consists in an activation of messages issued from glucose sensors of various organs.
These hormonal and neural signals modulate carbohydrate metabolism by controlling glucose fluxes, including endogenous production and the entrance of glucose into the cells. The translocation of glucose transporters (GLUT) is the prominent mechanism for the modulation of glucose transport across the cell membranes.9 Among those transporters, GLUT-1 is the predominant transporter for noninsulin-mediated glucose uptake (NIMGU) (Fig. 21-1).32 GLUT-2 regulates the flow of glucose across liver cell membranes. GLUT-4 is the main insulin-responsive glucose transporter and therefore modulates the insulin-mediated glucose uptake (IMGU) in adipose tissue, cardiac and skeletal muscles.
FIGURE 21-1
Insulin and glucose uptake by tissues in physiological conditions. Insulin promotes insulin-mediated glucose uptake (IMGU) in adipose tissues, skeletal and cardiac muscles by activating GLUT-4 transporters. Simultaneously, insulin activates GLUT-2 transporters in the liver, which decrease the endogenous glucose production. The global effect is a decrease in blood glucose level (insulin is a hypoglycemic hormone). As a consequence, noninsulin-mediated glucose uptake (NIMGU) by GLUT-1 transporters decreases. (Adapted with permission from Lena D, Kalfon P, Preiser JC, Ichai C. Glycemic control in the intensive care unit and during the postoperative period. Anesthesiology. February 2011;114(2):438-444.)
TOXICITY ASSOCIATED WITH HIGH GLUCOSE CONCENTRATIONS
Because glucose is the preferential substrate during critically ill conditions, stress hyperglycemia was considered for a long time as a beneficial response allowing an adequate provision of energy to tissues. However, in stress conditions, an overall massive glucose overload happens in NIMGU tissues under the influence of proinflammatory mediators, counterregulatory hormones, and hypoxia. A wide range of tissues, including hepatocytes, endothelial cells, neurons, nephrons, and immune cells may be susceptible to enhanced glucose toxicity as a result of acute illness. In these tissues, several deleterious effects have been associated with these high glucose concentrations in cells.1,9 Damages to mitochondrial proteins occur and the formation of reactive oxygen species (ROS) is increased as a consequence of the shift from glycolysis toward accessory metabolic pathways (pentose phosphate, hexosamines, polyols).10 Other effects of excess glucose concentrations include the exacerbation of inflammatory pathways, decreased complement activity, modifications in the innate immune system, impairment in endothelial and hepatic mitochondrial functions, abolishment of the ischemic preconditioning, and protein glycosylation. Acute complications attributed to stress hyperglycemia include renal failure, increased susceptibility to infections, polyneuropathy, and impaired microcirculation.1
MECHANISMS OF STRESS HYPERGLYCEMIA
Although sharing some similarities, the pathogenetic mechanisms of type 2 diabetes and stress hyperglycemia are different. In diabetes, the cause of hyperglycemia is a combination of insulin resistance and defective secretion of insulin by pancreatic β-cells. During stress hyperglycemia, complex interactions between counterregulatory hormones (catecholamines, growth hormone, and cortisol) and cytokines lead to an excessive hepatic glucose production and peripheral insulin resistance (Fig. 21-2). This highly complex interplay is largely variable over time.1,11
FIGURE 21-2
Glucose metabolism in stress hyperglycemia. Stress hyperglycemia is marked by increased whole-body glucose uptake in noninsulin-mediated glucose transport. Insulin-mediated glucose transport is reduced (insulin resistance). Intracellular nonoxidative glucose metabolism is also impaired. (Adapted with permission from Dungan KM, Braithwaite SS, Preiser JC. Stress hyperglycaemia. Lancet. May 23, 2009;373(9677):1798-1807.)
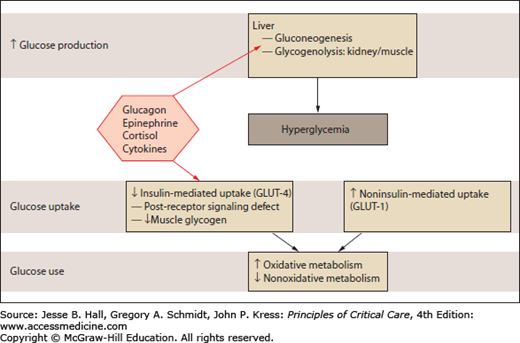
The stress-related increase in hepatic output of glucose results from gluconeogenesis and to a lesser extent from glycogenolysis. Gluconeogenesis is triggered to a larger extent by glucagon than by epinephrine and cortisol. Glycogenolysis is primarily triggered by catecholamines and perpetuated under the influence of epinephrine and cortisol. Tumor necrosis factor-α (TNFα) might promote gluconeogenesis by stimulating glucagon production. The increase in peripheral resistance is characterized by the inability of skeletal muscles and adipocytes to take up glucose, related to an alteration of insulin signaling and with a downregulation of GLUT-4 transporters (Fig. 21-1).
An increased glucose reabsorption or a decreased renal glucose clearance have also been reported and likely contribute to hyperglycemia in acute conditions.12 Surgical stress itself is an important trigger, via the induction of insulin resistance under the influence of cytokines and counterregulatory hormones. The degree of insulin resistance has been related to the magnitude and the duration of the surgical stress. The avoidance of hypothermia, excessive blood losses, prolonged preoperative fasting period, and prolonged immobilization synergize to reduce perioperative insulin resistance.







Full access? Get Clinical Tree


