118 Glomerulonephritis and Interstitial Nephritis
Over half of all critically ill patients develop some degree of acute kidney injury (AKI), and nearly 5% require renal replacement therapy (RRT). For those patients with severe AKI requiring. RRT, mortality can be as high as 70%, and up to 30% of surviving patients remain dialysis dependent.1–6 AKI may be a consequence of prerenal causes resulting in hypoperfusion of the kidneys, intrinsic renal causes, and postrenal or obstructive causes. In critically ill patients, the majority of AKI is related to ischemic or toxic acute tubular injury, which is treated supportively and is often reversible. AKI related to acute glomerulonephritis (GN) and acute interstitial nephritis (AIN) occurs in a smaller percentage of patients, but the incidence may be as high as 20% of all AKI.7 In addition to supportive care, initiation of correct treatment regimens is paramount for patient and renal survival. The focus in this chapter is on the renal causes of AKI, particularly GN and AIN.
 Glomerulonephritis
Glomerulonephritis
In GN, patients present with nephritic syndrome characterized by hematuria, proteinuria, AKI, edema, and hypertension.8 Hematuria may be microscopic or macroscopic, and urine sediment demonstrates dysmorphic red blood cells (RBC) and RBC casts. Urinary protein excretion typically exceeds 1 gram per day, and the degree of proteinuria can be rapidly assessed using a spot urine protein-to-creatinine ratio. In some instances, patients may have nephrotic-range proteinuria (>3 g/d) with associated clinical manifestations including edema, hypoalbuminemia, and hypercholesterolemia. Leukocyturia with or without white blood cell casts may be observed with GN of inflammatory origin.
In renal biopsy series of patients with unexplained AKI, the most common diagnoses included various forms of GN (pauci-immune GN, immunoglobulin [Ig]A nephropathy, postinfectious GN, lupus nephritis, anti–glomerular basement membrane [anti-GBM] disease) and AIN.7,9–11 Indeed, the third most common cause of end-stage kidney disease (ESKD) in the United States and Europe is GN.8 Distinguishing the type of GN with renal biopsy is critical for diagnosis as well as assessing the degree of acute versus chronic disease, which helps guide treatment and prognosis.
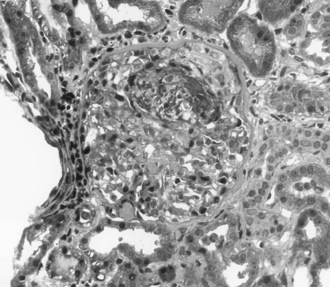
The most aggressive form of GN is described clinically as rapidly progressive glomerulonephritis (RPGN). Rather than a single disease entity, RPGN is the severe form of many of the glomerular diseases that are divided into renal limited etiologies and systemic diseases that involve the kidneys (Table 118-1). RPGN is defined as rapidly declining renal function, progressive oliguria, hematuria, proteinuria, and hypertension.8 Although many critically ill patients may have hematuria associated with infection or trauma, hematuria and AKI should always prompt consideration of acute GN. Renal ultrasound documents normal renal blood flow and normal to slightly enlarged kidneys. Renal biopsy reveals a high degree of glomerular injury with extensive crescent formation (Figure 118-1). Importantly, the transition from an acute cellular crescent to chronic, irreversible injury may occur rapidly over days. The presentation of a patient with RPGN constitutes a need for prompt diagnosis with early intervention and therapy to interrupt a natural progression to chronic renal failure. In adults, the most common cause of RPGN is pauci-immune GN associated with antineutrophil cytoplasmic antibodies (ANCA), and other common causes include Goodpasture’s syndrome (or anti-GBM disease) and immune-complex disease such as lupus nephritis.8,12 Immunohistology of the renal biopsy shows pauci-immune staining in ANCA-associated GN, linear IgG staining of the GBM in Goodpasture’s syndrome, and immune complex deposition in lupus nephritis, IgA nephropathy, and postinfectious GN.
TABLE118-1 Diseases Associated with Rapidly Progressive Glomerulonephritis and Pertinent Laboratory Studies
| Renal Limited | |
| IgA nephropathy | |
| Postinfectious glomerulonephritis | Low complement, streptococcal serologies, bacterial cultures |
| ANCA-associated glomerulonephritis (pauci-immune glomerulonephritis) | ANCA titers |
| Anti-GBM disease (Goodpasture’s syndrome) | Anti-GBM antibodies |
| Systemic Disorders | |
| Lupus nephritis | Low complement, ANA, dsDNA antibodies |
| ANCA-associated small-vessel vasculitis | ANCA titers |
| Anti-GBM disease | Anti-GBM antibodies |
| Henoch-Schönlein purpura | None |
| Cryoglobulinemic vasculitis | Low complement, cryoglobulins, hepatitis C serologies |
ANCA, antineutrophil cytoplasmic antibodies; ANA, antinuclear antibodies; dsDNA, double-stranded DNA; GBM, glomerular basement membrane; IgA, immunoglobulin A.
Pulmonary renal syndrome, characterized by RPGN and diffuse alveolar hemorrhage (DAH), often presents as a medical emergency requiring early aggressive treatment.13–15 It is associated with high mortality rates and rapid progression to ESKD if left untreated. Admission to the intensive care unit (ICU) and mortality are related to both the disease itself and infection. Patients often present with dyspnea, fever, cough, and hemoptysis, with chest radiography documenting pulmonary infiltrates. It may be difficult to distinguish from pneumonia, especially in patients without hemoptysis. Roughly 30% of patients with DAH do not present with hemoptysis. The presence of renal dysfunction and hematuria in patients presenting with these pulmonary symptoms should raise suspicion for a pulmonary renal syndrome. Although Goodpasture’s syndrome was first used in 1958 to describe patients presenting with pulmonary hemorrhage and GN,16 the most common cause of pulmonary renal syndrome is actually ANCA-associated small-vessel vasculitis.8 Goodpasture’s syndrome (also termed anti-GBM disease) now refers to the triad of DAH, RPGN, and the presence of anti-GBM antibodies and is the second most common cause of pulmonary renal syndrome. Much less common causes of pulmonary renal syndromes are SLE (systemic lupus erythematosus), thrombotic microangiopathies, and other systemic vasculitides.
Pauci-Immune Necrotizing Glomerulonephritis
Mortality of untreated disease is roughly 90% at 2 years following disease onset.17 However, systematic studies of different treatment regimens have led to significant progress in this field and improved patient outcomes.18 Treatment consists of pulse intravenous (IV) methylprednisolone followed by oral corticosteroids and IV cyclophosphamide.14,19–21 Even patients who are dialysis dependent on presentation often recover renal function with appropriate treatment. Poor prognostic indicators for patient and renal survival are the presence of DAH, severity of renal injury at diagnosis, degree of glomerular injury, extent of tubulointerstitial lesions on biopsy, and older age.22–26 Patients with DAH have a high mortality rate, and plasma exchange improves patient survival.17,27,28 For severe pulmonary disease, a few patients have been successfully treated with ECMO (extracorporeal membrane oxygenation).29,30 Additionally, patients with severe renal disease have an increased likelihood of renal recovery when treated with plasma exchange.23,25,31–33 With appropriate treatment, roughly 80% to 90% of patients achieve remission.17,21,27,34,35 Treatment resistance is more common in females, African Americans, and patients with severe renal disease. Relapse is more common in patients with anti-PR3 antibodies and involvement of the pulmonary and upper respiratory systems. The ANCA-associated small-vessel vasculitides follow a remitting and relapsing course, making long-term monitoring a key component to patient and kidney survival.
Anti-Glomerular Basement Membrane Glomerulonephritis
Goodpasture’s syndrome or anti-GBM disease presents as DAH and RPGN with evidence of anti-GBM antibodies on serologic testing. However, roughly 30% to 40% of patients present with renal limited disease without pulmonary involvement. It commonly affects Caucasians in a bimodal age distribution with peaks during the third and sixth decades.14,36–38 Renal biopsy shows linear deposition of antibodies, most commonly IgG and C3, along the GBM and glomerular crescent formation.
Untreated disease is highly fatal, and death is usually due to pulmonary hemorrhage or infection. Treatment with plasma exchange, cytotoxic agents, and corticosteroids was introduced in the 1970s, resulting in improved patient and renal survival.39 Plasma exchange is crucial for rapid clearance of anti-GBM antibodies40 and should be continued daily until antibodies are undetectable.38 Long-term outcomes are related to the degree of pulmonary compromise and renal dysfunction at presentation. With appropriate treatment, survival rates may exceed 90% for acute disease, but patients requiring RRT on initial presentation have lower survival rates.37,38,41 For those patients, only a very few recover renal function despite treatment with plasma exchange, corticosteroids, and cyclophosphamide. In contrast, those patients with creatinine (Cr) below 5.7 on presentation demonstrated 100% 1-year patient survival and 95% renal recovery in one study.37,41 In addition to dialysis dependence and elevated creatinine, predictors of poor renal outcome include oligoanuria, high anti-GBM antibody titers, and high percentage of glomeruli with crescent formation and extensive tubulointerstitial disease on renal biopsy.36,40,42,43 Although patient and renal survival is generally worse with anti-GBM disease than with ANCA-associated disease, late recurrence of anti-GBM disease is much rarer than recurrence of ANCA-associated disease.12,38
Both ANCA-associated vasculitis and anti-GBM disease are rare diseases, and interestingly, a subset of patients actually demonstrates both types of antibodies on serologic studies. Roughly 15% to 30% of patients with ANCA-associated disease also have anti-GBM antibodies, while only 5% to 10% of patients with anti-GBM antibodies also have detectable ANCA titers.12,37,38,44–46 Although outcome data are limited in this small group of patients, the outcomes of these patients may be better than patients with only anti-GBM antibodies.
Lupus Nephritis
Lupus nephritis occurs in 40% to 70% of patients with SLE and often occurs in the first 2 years following diagnosis.47–49 Less than 5% of patients present with RPGN or pulmonary renal syndrome. However, 10% to 20% of patients with lupus nephritis ultimately progress to ESKD. In addition to history and physical examination, evaluation includes analysis of urine sediment (because lupus nephritis may present as nephritic or nephrotic syndrome), assessment of proteinuria, complement levels, and serologies for ANA and anti-dsDNA antibodies. Cellular casts or proteinuria over 0.5 g/d is consistent with the diagnosis of lupus nephritis. Renal biopsy is critical for diagnosis, prognosis, and guiding treatment.
Renal biopsy is used to classify lupus nephritis into six categories: class I (minimal mesangial lupus GN), class II (mesangial proliferative lupus GN), class III (focal proliferative lupus GN), class IV (diffuse proliferative lupus GN), class V (membranous lupus GN), and class VI (advanced sclerosis).50–52 The most severe classes are the proliferative lesions of lupus nephritis (classes III and IV) and have poor renal survival without aggressive treatment. These classes often present with hematuria, proteinuria, hypertension, and AKI. Patients who present with RPGN are likely to have class IV lupus nephritis on renal biopsy. Sclerosing lupus nephritis is a chronic lesion that carries a poor prognosis.
Treatment of the more severe forms of lupus nephritis includes pulse methylprednisolone followed by oral corticosteroids and IV cyclophosphamide.48,53–57 Similar to treatment of pauci-immune GN, pulse IV cyclophosphamide is preferred over oral cyclophosphamide. Over 80% of patients respond to treatment.47,48 Importantly, about 5% to 10% of patients who require RRT initially recover enough renal function to become dialysis independent following treatment.49 Recent studies suggest that mycophenolate mofetil (MMF) is similar to cyclophosphamide in inducing remission; however, relapse appears more common in patients treated with MMF.58–62 Induction should be followed by maintenance therapy; the optimal maintenance regimen remains under intense investigation. Options for maintenance include additional cyclophosphamide, azathioprine, and MMF.48,57,63,64
Poor prognostic indicators at the beginning of treatment include male gender, African American race, severe hypertension, antiphospholipid syndrome (APS), and delayed initiation of immunosuppressive therapy. Following induction treatment, poor prognostic indicators are failure to achieve remission at 6 months and uncontrolled hypertension.48,49 Roughly one-third to half of patients will have relapse of disease. In some patients, recurrence of disease may be preceded by falling complement levels and rising anti-dsDNA titers. However, some patients with severe lupus nephritis have negative titers.47,65 Patients with only partial remission often recur sooner than patients with complete remission, and they are more likely to progress to ESKD.66 All patients with a history of lupus nephritis should be carefully monitored for recurrence of disease, and repeat renal biopsy is often needed to guide treatment decisions with relapsed disease.
Postinfectious Glomerulonephritis
Postinfectious glomerulonephritis (PIGN) presents as a classic nephritic syndrome occurring about 1 to 3 weeks after a group A β-hemolytic Streptococcus infection.67–69 It commonly occurs in children following a skin or pharyngeal infection. Although PIGN remains the most common cause of acute nephritic syndrome in the pediatric population in developing countries, the incidence of this disease has declined dramatically in the industrialized world. Children present with a classic nephritic syndrome with hematuria, proteinuria, hypertension, edema, and mild renal impairment. Severe hypertension with encephalopathy and seizures is uncommon and may require admission to the ICU.69–71 Laboratory findings demonstrate depressed complement levels (CH50 and C3) consistent with activation of the alternate complement cascade; levels return to normal by 8 to 12 weeks. Serologic studies may be used to confirm recent streptococcal infection, particularly with recent pharyngitis.69,72,73 Renal biopsy demonstrates endocapillary proliferation and granular deposition of immune complexes by immunohistology.67,72,74–76
The acute nephritic syndrome usually resolves in 7 days, and the prognosis of children with PIGN is excellent. However, roughly 10% to 20% of children have persistent urinary abnormalities including proteinuria and hematuria.68,69,73,77–79 Treatment is generally supportive with antihypertensives and diuretics as needed in the acute phase. Active infections should be treated, and prophylactic antibiotics are often indicated in endemic situations and for household contacts in regions with high prevalence of disease.
In contrast to children, outcomes for PIGN in adults in the industrialized world are much worse, particularly for patients with underlying chronic disease.67,68,80–82 PIGN can be associated with almost any infection, including most streptococcal and staphylococcal strains, gram-negative bacteria, mycobacteria, viruses, fungi, and parasites. Elderly patients often present with AKI, congestive heart failure, and nephrotic-range proteinuria. Up to half of these patients have underlying chronic diseases or risk factors including diabetes mellitus, liver disease or alcoholism, cancer, and IV drug use.80–83 Some patients demonstrate skin or pharyngeal infections, but many have other infections such as endocarditis and pulmonary infections. Streptococcal and staphylococcal infections account for only half of cases. Treatment consists of supportive care and eradication of infection. Although recent studies of adults with PIGN remain small, one-quarter to one-half of patients have persistent renal dysfunction, and as high as 15% may progress to ESKD.80–8284 In one small study, patients with underlying diabetic nephropathy had an extremely poor prognosis, with 81% progressing to ESKD.82
IgA Nephropathy
IgA nephropathy (IgAN) is an extremely common form of GN worldwide. However, IgAN is a renal-limited disease, with only 3% of patients presenting with AKI85 and most patients diagnosed in the outpatient setting. It commonly presents in the second or third decade of life and affects males more often than females.86 The majority of patients present with macroscopic or microscopic hematuria. Many patients have episodic hematuria, often associated with a concurrent upper respiratory tract or gastrointestinal infection. Patients may develop hypertension and varying degrees of proteinuria. Crescentic IgAN is associated with nephrotic-range proteinuria, severe hypertension, and rapidly declining renal function.87 No specific laboratory study to date can establish the diagnosis; renal biopsy is required. The extent of changes by light microscopy is variable, and the diagnosis is based on the demonstration of mesangial IgA deposits by immunohistology.
The long-term prognosis of patients with IgA nephropathy is highly variable, but many patients develop progressive renal failure. Between 15% and 40% of patients reach ESKD within 10 to 20 years of diagnosis.88,89 No consensus on the optimum treatment of IgAN is available owing to the lack of well-designed controlled trials. Progress is hampered by the fact that renal failure develops slowly over decades, and short clinical trials have limited usefulness.74,88,90,91 In all patients, hypertension should be aggressively treated with renin-angiotensin blockade. Patients with significant proteinuria and declining renal function may benefit from corticosteroids or immunosuppressive agents. Corticosteroids appear to reduce the risk of progression to ESKD and decrease proteinuria in selected patients.89,90,92 The small percentage of patients presenting with RPGN and crescentic GN are usually treated with pulse corticosteroids and cyclosphosphamide.87,88 Predictors of disease progression include renal dysfunction at diagnosis, significant proteinuria, hypertension, and evidence of chronic disease by renal biopsy.86,88,93,94


Full access? Get Clinical Tree


