Genetics of Migraine
Michel D. Ferrari
Joost Haan
Aarno Palotie
Anyone can experience an occasional migraine attack without necessarily being a migraine patient. Not the attack itself, but the repeated occurrence of attacks, is abnormal. In this respect, migraine is very similar to other paroxysmal disorders, such as epilepsy. Migraine often runs in families, suggesting that genetic factors, among others, influence the individual threshold for attacks. Next to genetic factors, other endogenous and exogenous factors modulate this threshold, and migraine is therefore considered a multifactorial disorder (47). Unraveling the genetic basis of migraine should improve our understanding of the pathogenesis of the disease, notably how, why, and when patients experience attacks. This improved insight into the mechanism of the onset of migraine attacks may promote the development of migraine-specific prophylactic drugs. In addition, it may help to establish an objective diagnostic test for (subtypes of) migraine (47). This chapter updates the genetic data on migraine without aura (MO), migraine with aura (MA), familial hemiplegic migraine (FHM), and related disorders.
FAMILY STUDIES
Transmission of migraine from parents to children was reported as early as the seventeenth century (200). Since that time, a large number of studies have reported a positive family history of migraine (reviewed in Russell [149]). Although a positive family history is characteristic of genetic traits, it is obviously not direct evidence for a genetic contribution because families often have a shared environment. Moreover, in very common traits such as migraine (lifetime prevalence of 16 to 21% in the general population (145,153), the chance of a positive family history increases by itself. Therefore, to provide evidence for a genetic contribution, a positive family history should include more stringent criteria than only affected first-degree relatives. In addition, the method of recording family history can also influence the result.
Despite the fact that familial aggregation of migraine is a well-recognized clinical observation and that many studies in the past have supported this empirical observation, there are few studies that meet modern methodologic standards. Apart from problems in collecting data (e.g., questionnaires, interviews), most studies lack validated diagnostic criteria, like those of the International Headache Society (IHS). Two population-based studies randomly selected individuals who were subsequently interviewed (152,162). Russell and Olesen (152) selected 4,000 (3,000 male and 1,000 female) 40-year-old individuals from the Danish Central Person Registry. On the basis of a questionnaire, these authors selected those with self-reported migraine and invited them for a headache interview and a physical and neurologic examination. Spouses and first-degree relatives were interviewed by phone and classified according to IHS criteria. This resulted in 270 probands with migraine: 126 had MO, 127 MA, and 17 had both types of migraine. Interestingly, a different pattern of familial risk was observed for MA and MO. Compared with the general population, the first-degree relatives of probands with MO had 1.9 times the risk for MO but only 1.4 times the risk for MA. On the other hand, first-degree relatives of probands with MA had almost 4 times the risk for MA but no increased risk for MO. The authors concluded that MO was caused by a combination of genetic and environmental factors, whereas MA was determined largely by genetic factors.
A population-based study by Stewart et al. (162) collected a random sample of households in Baltimore County, Maryland, using a random-digit dialing method. The survey was based on either a telephone interview or a self-administered questionnaire. Individuals with probable migraine were invited for a clinic visit for interview and general medical and neurologic examinations. This resulted in 73 migraine probands and 511 first-degree relatives. Probands were stratified into those having more disabling symptoms, missing work or school, and those who only rarely had to skip work or school because of migraine.
The risk for migraine was considerably higher among relatives of probands with disabling migraine (relative risk [RR] 2.17) compared to relatives of probands with minimal disability (RR 1.20). This risk was not influenced by the type of the proband’s migraine. An increased risk for both types of migraine was found in family members, regardless of whether the proband had MA or MO. The authors concluded that familial factors contribute to less than 50% of all migraine cases.
The risk for migraine was considerably higher among relatives of probands with disabling migraine (relative risk [RR] 2.17) compared to relatives of probands with minimal disability (RR 1.20). This risk was not influenced by the type of the proband’s migraine. An increased risk for both types of migraine was found in family members, regardless of whether the proband had MA or MO. The authors concluded that familial factors contribute to less than 50% of all migraine cases.
Noble-Topham et al. estimated the genetic load in familial MA, by comparing sibling risk, age at onset, and aura type in 54 MA probands categorized by family history of MA (125). Families with an MA proband were divided into families with MA in three generations (n = 15), two generations (n = 20), and one generation, so only the proband was affected (n = 19). The recurrence risk to siblings of probands was 2.7-fold higher in three generation compared with two-generation MA families and 4.8-fold higher in three-generation compared with one-generation MA families. MA probands from three-generation families were significantly younger than probands with no affected family members. The significant difference in genetic load and the earlier age at onset in the three-generation families is further evidence of a genetic basis for MA.
TWIN STUDIES
Several large, population-based twin studies have been published regarding the concordance of migraine in monozygotic twins (54,79,85,102,123,165,205). The older studies were performed before the introduction of the standard IHS criteria for the diagnosis of migraine, but have a trend similar to that of the newer studies, suggesting a higher concordance rate for migraine in monozygotic twins than in dizygotic twins in many populations (193).
Several studies in a Danish twin cohort (54,182) addressed most of the criticisms of older twin studies, because subjects were personally interviewed and IHS criteria were applied. The concordance rate for monozygotic twins indicated that genetic factors are a major contributor to the pathogenesis of both MO and MA. In a Finnish twin study, similar concordance rates were found (85). Mulder et al. (123) compared the prevalence and heritability of migraine across six of the countries that participate in the GenomEUtwin project, including a total of 29,717 twin pairs. The prevalence of migraine was ranging from 10 to 13% in Finland to 32 to 34% in Danish and Dutch women.
Another type of twin study focused on twins who had been raised apart. This setting provides an even more direct estimate of genetic influence because the effect of a shared environment is excluded. A drawback of these studies, however, is that the sample size is inevitably small because of the few individuals available. A study by Ziegler et al. (205) found that, in general, monozygotic twins had a higher concordance rate for migraine than dizygotic twins (total of 197 twin pairs) but did not find statistically significant evidence that monozygotic twins raised apart (23 pairs) would have a higher concordance rate for migraine than dizygotic twins raised apart (20 pairs). A small case-type study reported that two sets of monozygotic twins raised apart were concordant not only for migraine but also for the age at onset of the attacks (68). Svensson et al. studied a cohort of twins aged 42 to 81 years, including a subsample of 314 pairs reared apart and 364 matched control pairs reared together (165). They found no significant shared rearing environmental influences on migraine. The heritability of migraine was estimated at 38% for men and 48% for women. Interestingly, among monozygotic twins reared apart, those separated at 3 years of age or earlier were more similar for lifetime migraine than those separated later, and this was especially true for women. Svensson et al. suggested that family “resistance” in migraine is mainly caused by genetic factors, whereas environmental influences make family members different, not similar.
PREVALENCE OF MIGRAINE IN ETHNICALLY DIFFERENT POPULATIONS
The prevalence of migraine in African and Asian populations appears lower than in European and North American populations (134,176). These differences among racial groups may be caused by genetic rather than cultural or environmental factors because they persist in the United States. Stewart et al. (161) randomly selected and interviewed (by telephone) 12,328 individuals 18 to 65 years of age living in Baltimore County, Maryland. IHS criteria were used for the diagnosis of migraine. Migraine prevalence was significantly higher in whites than in African or Asian Americans. African Americans reported a higher level of headache pain but were less likely to report nausea and vomiting with their attacks compared to whites. By contrast, African Americans were less disabled by their attacks than were whites. Asian Americans and white Americans did not have significant differences in associated features.
MODE OF INHERITANCE
A number of attempts have been made to analyze the mode of inheritance of migraine. The early reports resulted in a number of conclusions, from autosomal recessive to autosomal dominant, with or without incomplete penetrance (reviewed in Russell [149]). A number of technical details, such as differences in diagnostic criteria and statistical analyses, contribute to the somewhat contradictory
conclusions. Because of the higher prevalence of migraine in women and the often-reported maternal transmission, X-linked and mitochondrial inheritance have been of special interest but were proved to be unlikely (116,138).
conclusions. Because of the higher prevalence of migraine in women and the often-reported maternal transmission, X-linked and mitochondrial inheritance have been of special interest but were proved to be unlikely (116,138).
Recent studies have been performed with up-to-date diagnostic criteria and statistical tools. Four studies published during the past 10 years have all used the IHS criteria for diagnosis, and all study subjects were individually interviewed. Three (83,151,183) of these four studies suggested a multifactorial inheritance. The other (116) suggested a recessive inheritance. A multifactorial inheritance was suggested regardless of whether the families were recruited from headache clinics (83,183) or from a population survey (151). Although various criticisms can be applied to all of these studies, there is a nearly 100% consistency of the results, suggesting a multifactorial mode of inheritance for migraine.
MOLECULAR STUDIES IN MIGRAINE
The search for genetic risk factors in paroxysmal diseases, such as migraine, is complicated by a number of clinical, genetic, and statistical problems. Major clinical issues are how to determine whether or not a person is affected and how to distinguish likely gene carriers from possible phenocopies. Another inherent problem is the high likelihood of locus heterogeneity. Although hard data are scanty, most linkage and association studies suggest that multiple genes contribute to the genetic susceptibility for migraine. Although early onset and severe clinical course are traditionally regarded as indicators of a genetic factor, it is unclear how one should deal with paroxysmal disorders. Are the number of attacks or their severity indicators of the presence of genetic risk factors, or are these merely a consequence of the frequency and intensity of the exposure to environmental triggers? The genetic strategy depends on the available patients, family material, and knowledge of likely candidate genes. When family material is abundant and candidate genes are scarce, a random genome screening for linkage is the method of choice. The preferred statistical method of analysis is debated, namely, parametric versus nonparametric, and the statistical thresholds that provide optimal distinction between linkage and background noise. Linkage findings may lead, after independent confirmation, to identification of positional candidate genes as opposed to the functional candidate genes that originate from insights into the biochemical pathways underlying the disease. The involvement of such functional candidates can be evaluated via functional assays and also by means of linkage tests in multiply affected families. A third, less commonly practiced method of identifying candidate genes for multifactorial disorders is localization of genes that cause rare Mendelian variants of that disorder. Such loci can then be evaluated as possible susceptibility loci, assuming that mutations that convey susceptibility to a complex disease are allelic to more serious gene defects leading to Mendelian segregation. Tentatively, one might call such candidate genes phenotypic candidates. Such rare variants (e.g., in the case of migraine FHM, or cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoen cephalopathy [CADASIL]; see below) usually have a clear inheritance pattern, and candidate loci can be identified by using regular logarithm of the odds (LOD) score analyses. With respect to functional candidates, one might object that their number is a priori not strictly defined and that different investigators may favor different functional candidates. In contrast, for phenotypic candidates the number of alternatives is usually limited. A fourth approach is investigating association with genetic markers. The rationale of such studies is that genetic markers, such as DNA polymorphisms, may occur in disequilibrium with genes, various alleles of which may lead to differences in the phenotype. Such linkage disequilibrium may have two possible reasons. Either the time that elapsed to separate the suspect genes from the tightly linked DNA polymorphism by recombination may not have been sufficient for the disequilibrium to disappear, or the marker allele itself influenced the phenotype to be studied.
FAMILIAL HEMIPLEGIC MIGRAINE
The only true molecular insight into migraine pathophysiology is provided by the isolation of the gene for a rare subtype of MA. FHM, a rare autosomal dominantly inherited subtype of MA, was first reported early in the twentieth century (26). A complete list of the early FHM literature is provided elsewhere (reviewed in Russell [149]). Attacks of FHM usually consist of a phase with hemiparesis in addition to at least two typical (visual) aura symptoms with a mean duration of 60 minutes, followed by a headache phase. FHM attacks resemble those of basilar migraine (61), and the headache and aura symptoms, apart from the hemiparesis, are identical to those of MA (173). Some patients have atypical attacks, either with a prolonged aura lasting up to 5 days or with signs of diffuse encephalopathy, expressed as confusion or coma, fever, and sometimes seizures. Atypical attacks are often the first symptoms of the disease and may be triggered by head trauma.
Patients with FHM and their unaffected relatives may also have attacks of MO or “nonhemiplegic” typical MA. Thomsen et al. studied the prevalence of MO and MA in probands with FHM and their first-degree relatives (174). Compared to the general population, FHM probands had virtually no increased risk of MO but a significantly increased risk of almost 8 times of MA. In total, 65% of FHM patients also have attacks of “normal” migraine: with aura 61%, without aura 18%, or with both types 21%. A similar pattern was seen among their first-degree relatives, who had no increased risk of MO, whereas the risk of
MA was significantly increased; 7.6 times in FHM-affected first-degree relatives and 2.4-times in non-FHM-affected first-degree relatives. These results suggest that the genetic abnormality resulting in FHM may also cause attacks with the symptomatology of MA. Thus, FHM and MA seem to be part of the same spectrum and FHM may be a good model to study the genetics of “normal” migraine.
MA was significantly increased; 7.6 times in FHM-affected first-degree relatives and 2.4-times in non-FHM-affected first-degree relatives. These results suggest that the genetic abnormality resulting in FHM may also cause attacks with the symptomatology of MA. Thus, FHM and MA seem to be part of the same spectrum and FHM may be a good model to study the genetics of “normal” migraine.
An increased risk might suggest that genes involved in FHM are candidate genes for MO and “nonhemiplegic” typical MA. Sporadic cases with FHM symptomatology exist (10,42,64,169), but they are not classified as FHM because this requires at least one affected first- or second-degree relative with identical attacks (63).
GENETIC STUDIES OF FHM
In 1993, linkage of FHM to chromosome 19p13 was reported (76), soon followed by proof of genetic heterogeneity, because only about 50% of the families appeared to be linked to this locus (77,132). Only a few clinical differences have been found between chromosome 19-linked and unlinked families (171), the most striking exception being cerebellar ataxia, which occurs in approximately 50% of the chromosome 19-linked but in none of the unlinked families (42,44,76,77,133). Another difference is that patients from chromosome 19-linked families are more likely to have attacks triggered by minor head trauma and associated with coma (42,170,171).
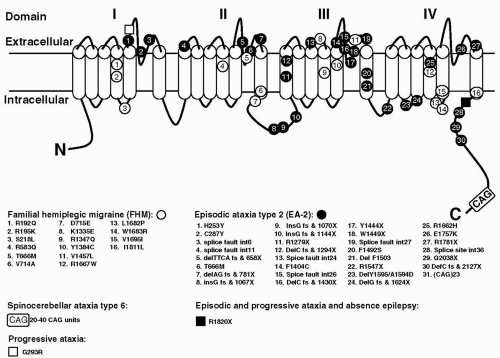 FIGURE 27-1. The CACNA1A gene with mutations. The α1A pore-forming subunit of P/Q-type voltage-gated calcium channels is located in the neuron membrane and contains four repeated domains. Each domain includes six membrane-spanning segments and a so-called P-loop between the fifth and sixth segments. Positions of mutations identified in this gene are given, including those identified in mice. |
The human gene originally designated CACNL1A439 has since been renamed CACNA1A (105). The gene encodes for the α1A subunit of voltage-gated P/Q-type neuronal calcium channels. The first missense mutations in the CACNA1A gene were found in FHM in 1996 (132). Since that time, several other mutations have been found (Fig. 27-1). There are mutations that cause pure FHM and others that are associated with FHM with ataxia. Ducros et al. (42) presented an extensive study on 21 FHM families
with eight different mutations. With a few exceptions, mutations were found in single, often small families. Only the T666M and the R385Q mutation were identified in 10 and 3 families, respectively. The T666M mutation appears to be the most common mutation worldwide, being found in several families from several countries (41,42,92,132,166,195). The finding of recurrent mutations allowed a comparison of all clinical features in mutation carriers. Of all mutation carriers, those with the T666M mutation showed the greatest frequency of hemiplegic migraine (98%), severe attacks with coma (50%), and nystagmus (86%). Thirteen subjects with mutations had no attacks of hemiplegic migraine, and some had MA (five subjects) or MO (one subject). A total of 66 atypical FHM attacks were reported in 44 mutation carriers. Some of these attacks were found to be prolonged (seven attacks in six patients), and most attacks were associated with encephalopathy (59 attacks in 38 patients).
with eight different mutations. With a few exceptions, mutations were found in single, often small families. Only the T666M and the R385Q mutation were identified in 10 and 3 families, respectively. The T666M mutation appears to be the most common mutation worldwide, being found in several families from several countries (41,42,92,132,166,195). The finding of recurrent mutations allowed a comparison of all clinical features in mutation carriers. Of all mutation carriers, those with the T666M mutation showed the greatest frequency of hemiplegic migraine (98%), severe attacks with coma (50%), and nystagmus (86%). Thirteen subjects with mutations had no attacks of hemiplegic migraine, and some had MA (five subjects) or MO (one subject). A total of 66 atypical FHM attacks were reported in 44 mutation carriers. Some of these attacks were found to be prolonged (seven attacks in six patients), and most attacks were associated with encephalopathy (59 attacks in 38 patients).
At the far end of the FHM CACNA1A spectrum is the clinical presentation of affected members of two families with an S218L missense mutation (48,93). These patients had recurrent atypical attacks, often triggered by trivial head trauma. The proband of the second family died after a period of coma following a symptom-free period of several hours after a mild head trauma. She had never experienced an attack of (hemiplegic) migraine. It appears that the S218L (and possibly other) CACNA1A calcium-channel mutations put persons at risk for uninhibited cerebral edema and fatal coma after minor head trauma.
Two studies point to the association of the CACNA1A gene with generalized epilepsy (25,78). Of four single-nucleotide polymorphisms (SNPs) and one microsatellite marker within the gene that were tested, one showed a significant association with epilepsy (25). However, because this SNP-8, located in exon 8, does not result in structural changes of the CACNA1A-encoded protein, it is unlikely that it is the causative mutation. Instead, SNP-8 is expected to be in linkage disequilibrium with such mutation. In another family, a missense mutation in the CACNA1A gene was associated with both episodic ataxia and epilepsy (78). These studies provided first evidence of the involvement of the CACNA1A locus in epilepsy in humans, which is of interest in view of the epileptic phenotype observed in CACNA1A mutant mice (see below).
Some years ago, two separate groups reported linkage of FHM to chromosome 1 (43,53). A North American group showed, in one large family, an LOD score of 3.04 at θ = 0.09 with marker D1S249 on chromosome 1q31 (53). Although suggestive evidence for linkage appeared to confirm this locus in one large Australian pedigree classified as “typical migraine” (103), the 1q31 locus was recently considered unlikely by the original investigators (90). Another group found linkage to chromosome 1q21-q23 in three FHM families (43), confirmed in other families (21).
One of the most important breakthroughs in the genetics of headache of the last 2 years is the identification of the second FHM gene (FHM2). This turned out to be the ATP1A2 gene, encoding the α2 subunit of sodium potassium pumps (30,107). Missense mutations were identified in two families (Fig. 27-2) and functional assays in cell lines suggested a loss of function as the underlying mechanism. Of interest, three subjects reported a history of seizures similar to migraine-triggered seizures observed in FHM1 patients. Two novel missense mutations in the ATP1A2 gene confirmed these results: one mutation was identified in a small family with pure FHM, the second in a large FHM family with benign familial infantile convulsions (BFIC), expanding the clinical phenotype associated with ATP1A2 mutations (189) (see Fig. 27-2). BFIC is a rare autosomal-dominant benign form of epilepsy, with strictly partial, nonfebrile, convulsions that begin at age 3 to 12 months and disappear after the first year. All BFIC persons tested had the missense mutation, but BFIC and FHM only partially cosegregated. It seems that migraine and epilepsy have partially overlapping mechanisms related to dysfunction of ion transport. Importantly, the discovery of the ATP1A2 gene opens up a new perspective on migraine mechanisms and therapies and resulted in a boost of genetic research (9,190).
Because some FHM families could not be linked to either chromosome 1 or 19, at least a third FHM gene must exist (43).
SPORADIC HEMIPLEGIC MIGRAINE AND THE CACNA1A GENE
The etiology of sporadic hemiplegic migraine (SHM), that is, hemiplegic migraine without affected family members, is unknown. Several studies searched for CACNA1A mutations in patients with SHM. In one study, two of three SHM patients with cerebellar signs had a mutation: a T666M mutation in a patient with cerebellar ataxia and a Y1384C mutation in a mentally retarded woman with recurrent prolonged attacks of hemiplegic migraine, coma, and seizures associated with permanent cerebellar ataxia and atrophy (184,185). Studies of small series of SHM patients showed mutations in the CACNA1A gene in two of three SHM patients with permanent cerebellar ataxia, but no mutations in eight patients without ataxia (19,41,42). In a recent study of 27 SHM patients, CACNA1A mutations were found in two of the patients (169). A patient carrying the T666M mutation was distinct from the others by showing permanent cerebellar ataxia and atrophy on computed tomography (CT) scan. Unfortunately, no DNA from the parents of this patient was available. Remarkably, the other mutation (R583Q) was found in a patient without cerebellar symptoms. This R583Q mutation had been reported previously in four families with FHM and associated ataxia (5,42).
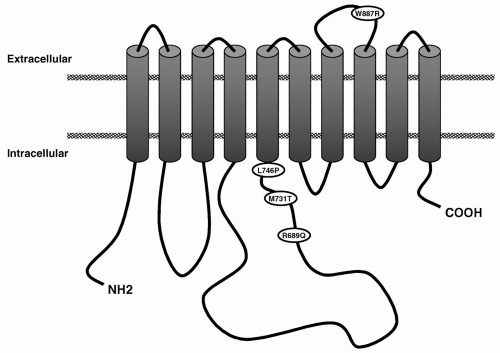 FIGURE 27-2. Predicted transmembrane model of Na+, K+-ATPase α2-subunit. The location of the various mutations is indicated. |
A study in a group of 105 SHM patients and their first-degree family members showed that more than half of the SHM patients had also nonhemiplegic MA attacks, and one third had additional MO attacks (175). First-degree family members of “pure SHM” patients had an increased risk of MA, whereas family members of all SHM patients together had an increased risk of both MO and MA. Another study showed that the clinical symptoms of SHM patients were more similar to FHM than MA (174).
EPISODIC ATAXIA TYPE 2 AND SPINOCEREBELLAR ATAXIA TYPE 6
Episodic ataxia type 2 (EA-2) is also referred to as acetazolamide-responsive paroxysmal cerebellar ataxia, paroxysmal vestibulocerebellar ataxia, or hereditary paroxysmal cerebellar ataxia (11). Onset is usually in childhood or early adulthood. Attacks are characterized by generalized ataxia lasting minutes to hours that may occur spontaneously or can be provoked by stress, exertion, alcohol, fever, and heat. Many patients also have interictal nystagmus and may display mild cerebellar atrophy. Up to 50% of patients experience migraine(like) symptoms during or in between attacks, or both. Magnetic resonance imaging (MRI) often reveals cerebellar atrophy (192). Attacks can be precipitated by emotional stress, exercise, or alcohol, and acetazolamide is often effective in preventing attacks. EA-2 appeared to be linked to the same interval on chromosome 19p as FHM (96,167,186,194) and was finally proved to be caused by mutations in the CACNA1A gene. Mutation analysis has revealed various different mutations in the CACNA1A calcium-channel gene in EA-2 families (see Fig. 27-1), including truncating mutations, resulting in premature stops and leading to putative aberrant CACNA1A proteins, and missense mutations (32,33,51,69,70,78,132,155,187). After several reports of new EA-2 mutations the total number is now over 20 (71,89,112,164). No CACNA1A mutation was identified in a family with episodic ataxia indistinguishable from EA-2, which suggests that EA-2 is a heterogeneous disorder and that other genes causing EA-2 should exist (65).
One of the EA-2 missense mutations caused additional generalized epilepsy (78), and another such mutation caused episodic weakness years before the onset of EA-2 attacks (69).
Spinocerebellar ataxia type 6 (SCA6) differs clinically from EA-2 by the presence of progressive rather than episodic ataxia (55). Six different cDNA isoforms of the CACN1A1 gene have been reported, of which three contained a 5-nucleotide insertion before the stop codon, resulting in a shift of the open reading frame in which the
CAG repeat is predicted to encode a polyglutamine stretch (204). Small triplet expansions of the intragenic CAG repeat, ranging from 21 to 30 repeat units, were observed in SCA6 patients, whereas normal chromosomes displayed 4 to 20 repeats (68,111,148,204). The CAG repeat length is inversely correlated with age at onset of ataxia (68, 111,148). Anticipation of the disease was observed clinically, but intergenerational allele size change was not observed, in contrast to other SCAs, which are also caused by CAG repeats. Two homozygotic cases of SCA6 have been reported. The first case could not demonstrate an unequivocal gene dosage effect on age at onset (111). The second case showed an earlier age of onset and more severe clinical manifestations than her sister, a heterozygote carrying an expanded allele with the same repeat length as the homozygote. SCA6 has been estimated to occur in 10% of Germans and 30% of Japanese with SCAs (111,148).
CAG repeat is predicted to encode a polyglutamine stretch (204). Small triplet expansions of the intragenic CAG repeat, ranging from 21 to 30 repeat units, were observed in SCA6 patients, whereas normal chromosomes displayed 4 to 20 repeats (68,111,148,204). The CAG repeat length is inversely correlated with age at onset of ataxia (68, 111,148). Anticipation of the disease was observed clinically, but intergenerational allele size change was not observed, in contrast to other SCAs, which are also caused by CAG repeats. Two homozygotic cases of SCA6 have been reported. The first case could not demonstrate an unequivocal gene dosage effect on age at onset (111). The second case showed an earlier age of onset and more severe clinical manifestations than her sister, a heterozygote carrying an expanded allele with the same repeat length as the homozygote. SCA6 has been estimated to occur in 10% of Germans and 30% of Japanese with SCAs (111,148).
FHM missense mutations seem by themselves (without CAG repeat expansions) sufficient to cause cerebellar ataxia (42,172,203). In contrast, two families with small CAG expansions showed paroxysmal symptoms (72). In one family with a clinical diagnosis of EA-2, a CAG23 repeat allele gave different interictal symptoms, ranging from nystagmus only to severe progressive cerebellar ataxia. In another family, initially classified as autosomal dominant cerebellar ataxia of unknown type, an intergenerational allele size change showed that a CAG20 allele was associated with an EA-2 phenotype and a CAG25 allele with progressive cerebellar ataxia.
FUNCTIONAL STUDIES OF CACNA1A MUTATIONS
Voltage-dependent Ca2+ channels are key protein structures in neuronal cell membranes (reviewed in Plomp et al. [141]). They transduce electrical signals into a cellular influx of Ca2+, which acts as a second messenger in many processes such as regulation of excitability, transmitter release, gene regulation, and axon growth. Ca2+ channels consist of a pore-forming, voltage-sensing α1-subunit, and auxiliary β-, α2 δ-, and γ-subunits. Several subtypes of heteromultimeric channels exist, which can be differentiated by pharmacologic and electrophysiologic criteria (i.e., N-, P/Q-, L-, R-, and T-types). The α1-subunit consists of four homologous repeats (I to IV), each with six transmembrane segments (S1 to S6; see Fig. 27-1). The so-called P-loop between S5 and S6 is believed to form the inner lining of the ion pore. The S4 segments have been shown to be involved in voltage sensing.
The CACNA1A gene encodes for the pore-forming subunit, α1A. Alternative splicing of CACNA1A yields either P- or Q-type channels (8). P/Q-type Ca2+ channels are widely expressed in the central nervous system (CNS), especially in the cerebellum. In cell bodies, P/Q-type Ca2+ channels play a role in excitability, presumably via Ca2+-dependent K+ channels. Furthermore, Ca2+ influx stimulates intracellular signaling pathways, mostly involving kinases, which can influence gene expression, for example. During development, P/Q-type Ca2+ channels appear to participate in the process of neurite initiation. At mature synaptic terminals, their main function is to mediate transmitter secretion by allowing Ca2+ to stimulate the release machinery complex at so-called active zones, resulting in exocytosis of synaptic vesicles. The α1A-subunit contains defined sites that interact with specific presynaptic proteins (see Fig. 27-1), presumably required for targeted presynaptic localization and specific presynaptic function of P/Q-type Ca2+ channels. Other specific sites for interaction with β-subunits and G-proteins have been identified.
Related posts:
 Principles of Clinical Pharmacology, Randomized Controlled Clinical Trials, and Evidence-Based Medicine in Headache
Principles of Clinical Pharmacology, Randomized Controlled Clinical Trials, and Evidence-Based Medicine in Headache
 Nitric Oxide
Nitric Oxide
 Human Models of the Headaches
Human Models of the Headaches
 Channelopathies and Their Possible Relation to Migraines
Channelopathies and Their Possible Relation to Migraines
 Autonomic Dysfunction in Migraines
Autonomic Dysfunction in Migraines
 Psychological and Behavioral Treatments of Migraines
Psychological and Behavioral Treatments of Migraines

Full access? Get Clinical Tree


