169 General Principles of Pharmacokinetics and Pharmacodynamics
Pharmacokinetics and pharmacodynamics describe, respectively, the amount of drug in the body at a given time and the pharmacologic effects caused by the drug.1 Pharmacokinetics describes the movement of a drug into, within, and out of the body over time, whereas pharmacodynamics explains the effects the drug has on the body that result in a clinical response. A general understanding of pharmacokinetic parameters such as clearance, volume of distribution, half-life, steady state, and absorption, along with pharmacodynamic principles such as receptor theory, potency, affinity, tolerance, and minimum effective concentration greatly enhances the clinician’s ability to make informed choices in the treatment of the critically ill patient.
 General Principles of Pharmacokinetics
General Principles of Pharmacokinetics
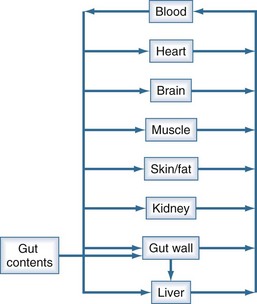
Clearance, volume of distribution, half-life, and bioavailability are four pharmacokinetic parameters that allow the clinician to better estimate dosing requirements. If the concentration of a drug in an easily assessable sampled fluid (e.g., plasma, urine, saliva) correlates well with the pharmacologic response (therapeutic or toxic) to the drug, then the application of pharmacokinetics in dosing is likely to be beneficial.2 Usually the concentration of a drug cannot be measured at the exact site of action (e.g., a receptor on the cell surface), so it is necessary that there be a predictable relationship between the concentration that is measurable and the concentration at the site of the effect.3,4 These concentrations do not have to be equal, but they should reflect a similar direction and magnitude of change over time (Figure 169-1).
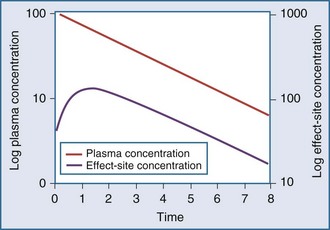
Measurement of the relationship between drug concentration and therapeutic or toxic response in a large number of patients allows development of a therapeutic range or target concentration for that drug (Figure 169-2).5–10 Table 169-1 lists a number of drugs commonly used in the ICU for which therapeutic ranges have been established and for which therapeutic drug monitoring is often recommended.11,12 Critically ill patients have a multitude of host factors (e.g., hemodynamic status, decreased organ function, nutritional status, concurrent disease states) that increase the likelihood that individualized drug dosing based on individualized pharmacokinetic assessment will be beneficial (Figure 169-3).13–16 There can be gender-related differences in both pharmacokinetic and pharmacodynamic responses.17–19 Individual chapters in this text are devoted to many of these agents and their adjustments for dosage in patients with renal or hepatic failure.
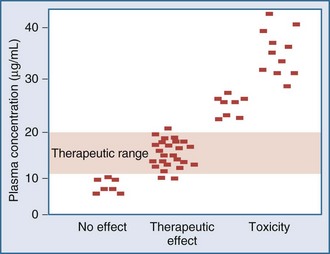
TABLE 169-1 Therapeutic Ranges of Drugs Commonly Used in Critical Care
| Drug | Therapeutic Range |
|---|---|
| Amikacin | Trough < 5 µg/mL |
| Peak < 30 µg/mL | |
| Cyclosporine | Whole blood, 150 ng/mL |
| Digoxin | 0.50-2.0 ng/mL |
| Gentamicin* | Trough < 2 µg/mL |
| Peak < 10 µg/mL | |
| Lidocaine | 1.5-5 µg/mL |
| Phenytoin | 10-20 µg/mL |
| Quinidine | 2-5 µg/mL |
| Theophylline | 10-20 µg/mL |
| Tobramycin* | Trough < 2 µg/mL |
| Peak < 10 µg/mL | |
| Vancomycin† | Trough < 5 µg/mL |
| Peak < 30 µg/mL |
* Once daily aminoglycoside dosing may result in different therapeutic ranges.
† Vancomycin concentrations are currently focusing on higher peak concentrations, and practice varies considerably between sites.
Pharmacokinetic Models
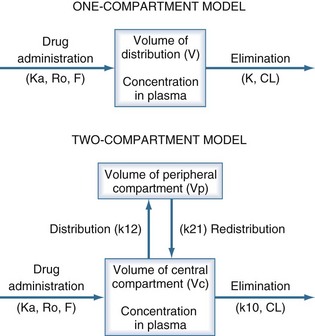
The pharmacokinetic concepts of clearance, volume of distribution, half-life, and bioavailability are based on physiologic principles.20 The physiologic processes governing these concepts are enormously complex, and many simplifying assumptions must be made before the mathematics describing drug concentrations become tractable. Although sophisticated computer modeling approaches are available in research settings, most of the clinically useful pharmacokinetic equations are based on one- or two-compartment models (Figure 169-4).21
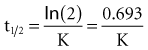
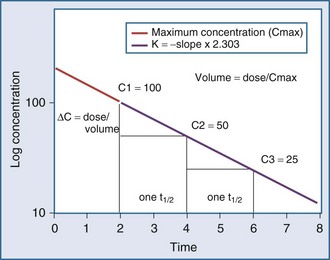
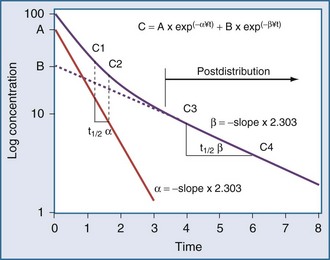
Specifically, t1/2 defines the length of time it takes for the drug concentration to decrease by one-half. In a linear pharmacokinetic system with first-order elimination, the t1/2 is a constant, and it takes the same amount of time for the concentration to fall from 100 to 50 arbitrary units as it does to decline from 50 to 25 arbitrary units (Figure 169-5).
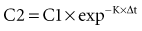
where Δt is the time elapsed between the measurement of two concentrations, C1 and C2. It is the properties of this equation that give rise to the familiar exponentially decreasing concentration-time curve, which becomes linear when plotted on semilog coordinates.
The human body is not a single well-stirred compartment, and it is amazing that such a simple mathematical model can be so useful in the clinical setting. If the body is conceptualized as consisting of individual tissues and organs, the same mathematical treatment can be applied. The concept of the volume of distribution has to be somewhat modified to recognize not only the physical size of the organ or tissue, but also the fact that drugs accumulate to differing degrees in different tissue spaces.22,23 For example, lipophilic drugs have a high affinity for adipose tissue, and this property is reflected by a large partition coefficient (R). The time constant associated with each tissue is a function of the rate of blood flow to that tissue (Q), the physical volume of the tissue (VT), and the partition coefficient. The time constant determines the rate at which equilibrium is reached. As a result, it is possible to construct a set of exponential equations with a time constant unique to each tissue:
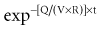
There is a branch of pharmacokinetics known as physiologically based pharmacokinetic modeling that uses blood flows, organ volumes, and partition coefficients to characterize concentration-time profiles.24–27 According to this approach for pharmacokinetic modeling, each tissue space ultimately contributes to the venous pool (Figure 169-6). However, the overall shape of the concentration-time profile in the venous blood is controlled not by the number of tissue spaces or their effective volumes, but by their time constants. Tissues with similar time constants, Q/(V × R), produce similar drug profiles in their venous outflows and appear as a single exponent in pooled venous blood. Practically speaking, many tissues and organs reach equilibrium over similar time frames, and often no more than two distinct time constants are observed. Therefore, this situation can be described adequately by a two-compartment model, characterized by a rapidly distributing central compartment and a more slowly equilibrating peripheral compartment (Figure 169-7). The equation describing the concentration-time profile for the two-compartment model is:
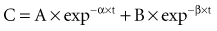
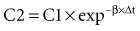
in which β replaces K, can still be used to predict concentrations, as long as both C1 and C2 are in the postdistributive phase. This sum-of-exponentials approach can be extended to three-compartment or even more complex models, but it is difficult to obtain all the concentrations needed to characterize each exponent.
Clearance
Clearance (CL) is a primary pharmacokinetic parameter that measures the ability of the body to eliminate a drug.28,29 It is often stated that clearance is the volume of blood (plasma) that is completely cleared of drug per unit time. Although this is one way to define clearance, it does not capture the relationship between drug clearance (mL/min) and the rate of drug elimination (mg/h). In pharmacokinetics, the general concept of clearance is defined as the rate of elimination relative to the concentration. In a first-order pharmacokinetic system, the rate of elimination is proportional to the drug concentration, and clearance is this proportionality constant:
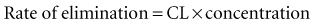
Clearance is clinically useful because it can be related directly to the organ of elimination. We can talk about renal clearance, hepatic clearance, or biliary clearance, and the sum of each of the individual clearances is the total body clearance.30,31 The immediate clinical consequence is the ability to adjust doses in response to changes in specific organ function. For example, a patient with developing renal failure is likely to require a reduction of the dose of a drug that is eliminated by the kidney, but not necessarily a reduction of the dose of a drug that is eliminated by the liver.32 If the clearance of a drug is known to be 50% renal and 50% hepatic, and renal function is decreased by 50%, it is necessary to reduce the dose by only 25% to maintain the same concentration.
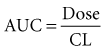
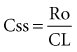
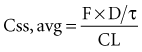
Volume of Distribution
The volume of distribution (V) is another primary pharmacokinetic parameter that is useful in determining the change in drug concentration for a given dose.33 After an intravenous bolus dose in a one-compartment pharmacokinetic model, the change in concentration (ΔC) between Cmax and the concentration immediately before the dose is administered is a function of the dose (D) and the volume of distribution (V):
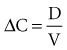
This equation is useful for predicting both the concentration after a first bolus dose and the increase in concentration at any point in time after a bolus dose. If a concentration before administration of a bolus dose is known or can be estimated, the equation can be used to predict the increase in concentration after the dose is administered (see Figure 169-5). It is important to recognize that the calculated value of ΔC must be added onto the pre-dose concentration to estimate the Cmax after the bolus dose. This equation is also useful for estimating the dose needed to reach a given concentration. If it is known that the volume of distribution is 0.45 L/kg, and a Cmax of 10 mg/L is desired after the loading dose, the dose is estimated to be 10 mg/L × 0.45 L/kg = 4.5 mg/kg. It is not necessary to have a steady-state condition to use this equation, a fact that makes it very useful in critical care.
The volume of distribution increases over time until a distribution equilibrium is reached among all compartments. This is the largest value for the volume of distribution. The fact that distribution equilibrium has occurred can be determined from a log-concentration versus time plot (see Figure 169-7). The curve becomes log-linear when the rate of drug entry into each peripheral compartment equals the rate of exit from each compartment. Because it is often calculated using the clearance and the β or terminal elimination half-life, this volume is often called Vβ:
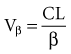
Half-Life
The half-life (t1/2) is a pharmacokinetic parameter defined as the length of time it takes to reduce the drug concentration by half (see Figure 169-5).33 The half-life is referred to as a secondary parameter because it is a function of the two primary parameters, clearance and volume of distribution:
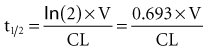
In a one-compartment system with constant clearance and volume of distribution, drug half-life also is constant. However, in a multicompartment model, the volume of distribution increases over time as drug equilibrates into tissue compartments until Vβ is reached. According to the previous equation, the half-life also increases over time and eventually reaches a maximum at t1/2β (see Figure 169-7).
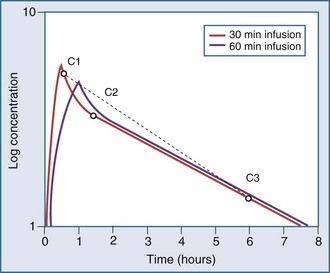
Most drugs have a rapid distribution phase that could be detected if concentrations were measured frequently enough. Aminoglycosides again are a good illustrative example of this concept, because they have a rapid, although not instantaneous, distribution phase (Figure 169-8). With a distribution phase half-life of 5 to 10 minutes, it would take approximately 25 to 50 minutes before the log-linear elimination phase could be observed. It is this distribution process that is the basis for the recommendation to wait approximately 1 hour after the end of an infusion before sampling blood to measure the aminoglycoside concentration. If a blood sample is obtained before this time, the drug still will be in the distribution phase, and the concentration measured will lead to underestimation of the drug half-life. In addition, slowly equilibrating compartments have been demonstrated when aminoglycoside concentrations are measured during washout.34 Aminoglycosides are usually dosed frequently enough so that the slowly equilibrating compartment is not detected.
< div class='tao-gold-member'>
Related posts:



Full access? Get Clinical Tree


