Optimal management of fluids, electrolytes, and acid–base status in critically ill patients requires a general understanding of their normal composition and regulation. Disease processes, trauma, and surgery can all affect the manner by which the body controls its fluid balance and electrolytes.
I. FLUID COMPARTMENTS
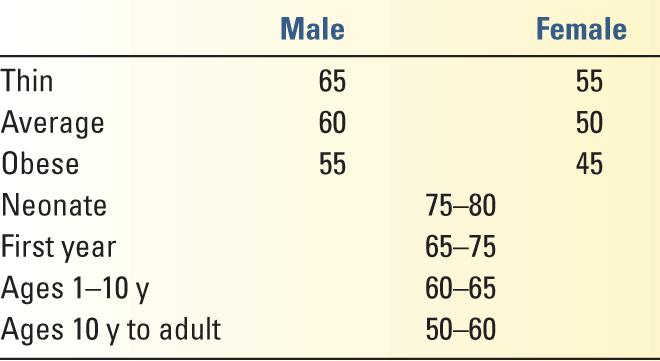
A. The Total Body Water (TBW) ranges from 50% to 70% of the body mass and is determined by lean body mass, gender, and age (Table 8.1). There is an inverse relationship between TBW and percentage body fat due to the low water content of adipose tissues.
B. Compartments of TBW
1. The intracellular compartment is approximately 66% of TBW (approximately 40% of body mass).
2. The extracellular compartment is approximately 34% of TBW (approximately 20% of body mass) and can be further divided into the following:
a. the intravascular compartment, composed of plasma. It is approximately 5% of total body mass.
b. the extravascular compartment, composed of interstitial fluid, lymph, bone fluid, fluids of the various body cavities, and mucosal/secretory fluids. The extravascular compartment represents approximately 15% of total body mass.
c. the third space, an ill-defined space invoked to account for otherwise inexplicable fluid losses in the perioperative period or in critically ill patients. It may be considered “nonfunctional” interstitial fluid that cannot equilibrate with the circulating blood volume. However, many experts doubt the existence of a discrete third space.
C. Ionic Composition of the Fluid Compartments. Various physiologic terms describe the concentrations of ions in a solution:
1. Molarity: Moles of solute per liter of solution
2. Molality: Moles of solute per kilogram of solvent
3. Osmolarity: Osmoles per liter of solution. The number of osmoles is determined by multiplying the number of moles of solute by the number of freely dissociated particles from one molecule of solute. For example, 1 mole of NaCl will yield 2 osmoles (Osm) in solution.
4. Osmolality: Osmoles per kilogram of solvent
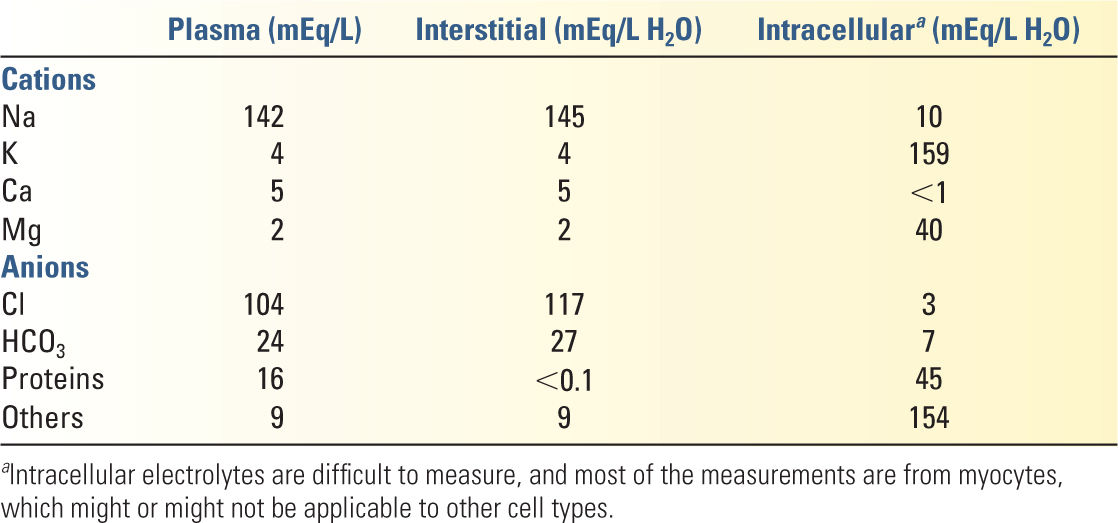
5. Electrolytes in physiology are generally described in terms of milliequivalents per liter (mEq/L). The fluids of each compartment are electrically neutral. Average concentrations of each electrolyte in various compartments are given in Table 8.2.
D. Movement of Water in the Body
1. Water is generally readily permeable through cell membranes and moves freely throughout the different fluid compartments. Movement of water is largely determined by osmotic pressure and hydrostatic pressure. The osmotic pressure depends on the number of osmotically active molecules in solution.
2. The movement of water between extracellular interstitial and intravascular compartments is described by Starling’s equation:
Qf = Kf [(pc − Pi) − σ(pic − pii)]
where Qf is the fluid flux across the capillary membrane; Kf is a constant reflecting the permeability of capillaries to water and their surface area; Pc and Pi are the hydrostatic pressures in the capillary and the interstitium, respectively; σ is the reflection coefficient (see below); and pic and pii are the colloid osmotic pressures in the capillary and interstitium, respectively.
a. Large, negatively charged intravascular proteins to which vascular membranes are impermeable are responsible for the osmotic pressure gradient between intravascular and interstitial compartments. This component of osmotic pressure, known as the oncotic pressure or colloid osmotic pressure, contributes a small amount to the total osmotic pressure of the fluids. The positive ions that are associated with the negatively charged proteins also contribute to the osmotic pressure. Albumin is the predominant type of protein responsible for the oncotic pressure, accounting for approximately two-thirds of the total oncotic pressure. Cells do not contribute to the oncotic pressure.
b. The reflection coefficient (σ) describes the permeability of plasma proteins for a specific capillary membrane. Its value ranges from 0 (completely permeable) to 1 (impermeable) and varies in different disease states; it is approximately 0.7 in healthy tissues.
c. Edema occurs when the rate of interstitial fluid accumulation is greater than the rate of removal of interstitial fluids by the lymphatic system.
II. FLUID DEFICITS AND REPLACEMENT THERAPY
A. Fluid Volume Deficits and appropriate fluid therapy are dependent on the source and type of fluid loss. Because all membranes are permeable to water, fluid deficits in one compartment will affect all other compartments. Fluid losses can be broadly classified on the basis of the initial source of loss. Fluid losses also lead to electrolyte abnormalities.
1. Intracellular fluid (ICF) compartment deficits arise from free water loss.
a. Sources of free water loss include the following:
1. Insensible losses through skin and respiratory tract
2. Renal losses secondary to inability to recollect water, such as in neurogenic or nephrogenic diabetes insipidus (DI; see Section III.C)
b. With free water loss, both intracellular and extracellular volumes decrease in proportion to their volumes in the body; therefore, two-thirds of the loss will be intracellular. Similarly, free water replacement will be distributed in proportion to their volumes in the body, and only one-third of the administered free water will end up in the extracellular space and even less in the intravascular space.
c. Therapy includes replacement of water with either hypotonic saline (5% dextrose in water with 0.45% sodium chloride solution) or free water (5% dextrose in water). Electrolytes (especially sodium) must be monitored with therapy.
2. Extracellular fluid (ECF) compartment deficit
a. In general, ECF losses are isotonic. Losses may occur in each compartment, and fluid losses from one compartment are rapidly reflected in others.
b. Clinical manifestations of ECF losses are notoriously variable. Although textbooks routinely refer to clinical findings associated with quantified losses (3%–5% losses leading to dry mucous membranes and oliguria to >20% losses leading to circulatory collapse), these signs are frequently unreliable. See the discussion on intravascular fluid deficit below.
c. Causes of ECF losses include blood loss, vomiting, diarrhea, and “third spacing.”
1. Replacement of ECF volume loss usually requires isotonic salt solutions. Volumes required to replace ECF deficits can vary considerably among patients on the basis of the inciting event and comorbid processes.
3. Intravascular fluid (plasma volume) deficit
a. Intravascular volume deficits will lead to interstitial fluid depletion as the two compartments equilibrate. It should be noted that with acute blood loss, laboratory values such as hemoglobin levels are not accurate indices of the severity of blood loss till equilibration with the ECF has taken place.
b. The clinical utility of classifications based on percentage volume loss and clinical manifestations (such as the ATLS classification of hemorrhage) is questionable, as signs of bleeding, including tachycardia, diaphoresis, and so forth, are extremely variable. Perhaps the most relevant clinical feature associated with intravascular volume loss is orthostatic hypotension (or orthostatic tachycardia) in a relatively stable patient. In an unstable patient, clinicians should use a combination of noninvasive or invasive methods to assess blood volume and “fluid responsiveness,” the ability of volume administration to increase cardiac output significantly. For a discussion of static and dynamic parameters predicting fluid responsiveness, see Chapters 1 and 3. Patients should be resuscitated early and aggressively—however, reliance on static numbers (e.g., 30 mL/kg crystalloid) or goals (e.g., give fluids till CVP is 8–12 mmHg) should be avoided in favor of frequent reassessment of both parameters of fluid responsiveness and global/regional perfusion indices.
B. Fluid Replacement Therapy (see Chapter 33)
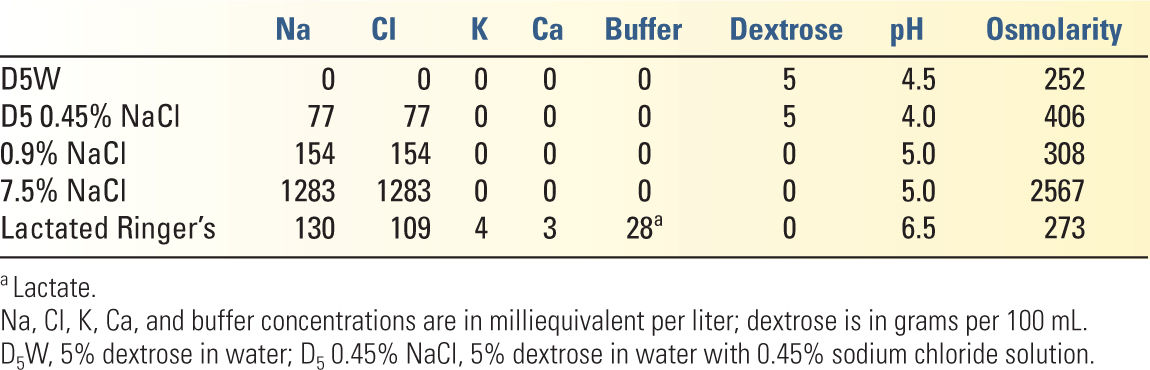
1. Crystalloid solutions (see Table 8.3 for the composition of commonly used crystalloids)
a. Maintenance fluids are used to replace constitutive losses of fluids and electrolytes via urine, sweat, respiration, and gastrointestinal (GI) tract.
1. Insensible water losses include normal losses by skin and lungs, and total approximately 600 to 800 mL/d. Sensible losses of water include losses from the kidneys and GI tract. The obligate minimal urine output is 0.3 mL/kg/h and the average urine output is 1 mL/kg/h in an average 70-kg person (approximately 1,700 mL/d).
2. General guidelines for hourly maintenance fluid replacement based on body weight (note that these guidelines should be tempered by frequent assessment of the patient’s volume status):
a. 0 to 10 kg: 4 mL/kg/h
b. 11 to 20 kg: 40 mL + 2 mL/kg/h for each kilogram above 10 kg
c. >20 kg: 60 mL + 1 mL/kg/h for each kilogram above 20 kg
3. Maintenance fluid composition. In general, hypotonic maintenance fluids are used to replace insensible losses. Additional losses from other sources are often present in critically ill patients (e.g., through drains, fistulas, etc.) and require isotonic fluid repletion.
b. For ECF repletion and intravascular fluid deficit (blood loss), isotonic solutions are used. The goal of replacement fluid therapy is to correct abnormalities in volume status or serum electrolytes. Because electrolytes are permeable through capillary membranes, crystalloids will rapidly redistribute from the intravascular compartment throughout the entire ECF, in the normal distribution of 75% extravascular to 25% intravascular.
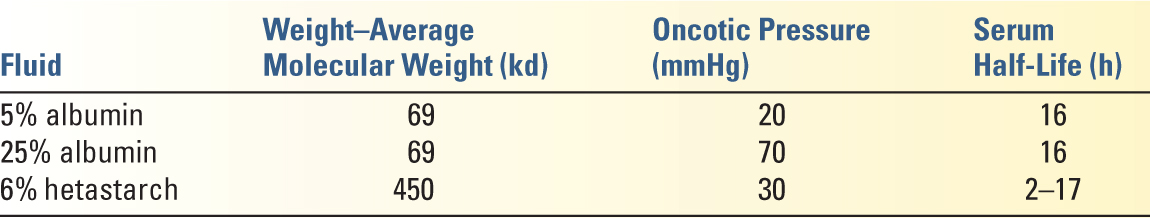
2. Colloid solutions (see Table 8.4 for the composition of commonly used colloids). Colloid solutions are most commonly used for intravascular volume expansion. Unlike crystalloid solutions, the colloid elements do not freely cross-intact capillary membranes and therefore do not redistribute as readily into the entire ECF compartment in the absence of a severe capillary leak state. Colloids have been thought to have a number of important potential advantages over crystalloids, although clinical evidence for these advantages is weak.
a. More effective volume expansion and less edema formation
b. Better at maintaining endothelial integrity
c. Improved microcirculation
d. Less immunomodulation
e. The chief disadvantage of colloids is their significantly higher cost compared with crystalloids (see below for the harmful effects specific to the hydroxyethyl starches).
3. Transfusions of blood and blood components are important for maintaining the oxygen-carrying capacity of blood and for coagulation. A detailed discussion of transfusions as a part of fluid management can be found in Chapter 33.
a. Transfusion should be restricted in critically ill patients who are not actively bleeding and do not have evidence of ongoing myocardial ischemia.
b. Patients needing massive transfusion should be transfused early with a 1:1:1 ratio of PRBCs to FFP to platelets for the best outcomes (see Chapter 33).
4. Clinical use: Crystalloids versus Colloids. The controversy over the ideal resuscitation fluid is an old one and is unlikely to be definitively answered in the near future. Nonetheless, in the last 3 to 4 years a number of large, well-designed trials have provided some guidelines for evidence-based fluid resuscitation—the recommendations below are based on our interpretation of these trials.
a. The initial choice of resuscitation fluid should almost always be a crystalloid. In spite of the expected advantages of colloids at volume expansion, the data shows only a 30% to 40% decrease in total volumes infused with colloid-based resuscitation compared with crystalloids (rather than the expected threefold difference), over days to weeks.
b. Lactated ringer’s (LR, or a balanced crystalloid) may be preferred in most instances to normal saline, even in the presence of renal dysfunction. With large-volume resuscitation, NaCl is associated with greater hyperchloremic acidosis, which may actually cause more hyperkalemia in patients with renal dysfunction than LR. In addition, accumulating evidence suggests that excessive chloride administration may be associated with worse renal outcomes, and perhaps worse mortality. One exception to this occurs in patients with neurologic injury, where resuscitation with relatively hypotonic fluids like LR is not recommended.
c. Hydroxyethyl starches (HES) are contraindicated in patients with severe sepsis and septic shock. There is high-quality evidence linking HES solutions and worse renal injury and mortality in patients with sepsis. It is probably prudent to avoid HES solutions in critically ill patients altogether.
d. Albumin is safe in most critically ill patients (with the exception of those with head injury, in whom it worsens mortality). The administration of albumin is not associated with improved outcomes in the broad population of critically ill patients. However, there are some groups of patients in whom albumin should be considered:
1. Patients with liver disease and cirrhosis undergoing large-volume paracentesis
2. Patients with septic shock and hypoalbuminemia. A recent study showed that keeping serum albumin levels >3 g/dL was associated with a mortality benefit in patients with septic shock (subgroup analysis of the ALBIOS trial).
III. ELECTROLYTES AND ELECTROLYTE ABNORMALITIES: SODIUM
A. The normal range of serum (plasma) sodium concentration is 136 to 145 mEq/L. Abnormalities in serum sodium suggest abnormalities in both water and sodium balance.
B. Hyponatremia is defined by a serum sodium of <136 mEq/L. Severe hyponatremia can cause central nervous system (CNS) and cardiac abnormalities, such as seizures and dysrhythmias. Hyponatremia can be classified on the basis of concomitant plasma tonicity.
1. Isotonic hyponatremia (approximately 290 mOsm/kg H2O) occurs when there are elevated levels of other ECF constituents such as protein and lipids. This form of hyponatremia is also known as pseudohyponatremia and is an artifact of measurement due to lipid and protein displacement of volume for a given volume of plasma. Therapy for this hyponatremia is not required.
2. Hypertonic hyponatremia is due to the movement of intracellular water into the ECF compartment under the influence of osmotically active substances (e.g., glucose, mannitol) with consequent dilution of ECF sodium. A common example of hypertonic hyponatremia is seen with hyperglycemia, which can decrease serum sodium concentration by approximately 1.6 mEq/L for each 100 mg/dL of blood glucose. Removal of the osmotically active etiologic substance and restoration of volume are goals of therapy.
3. Hypotonic hyponatremia is the most common form of hyponatremia. This type of hyponatremia is due to higher TBW relative to total body sodium. Hypotonic hyponatremia is further classified on the basis of the ECF volume status (hypovolemic, hypervolemic, and isovolemic). In all three scenarios, the extracellular compartment volume status does not always correlate with the intravascular or effective arterial volume status.
a. Hypovolemic hypotonic hyponatremia can result from renal or nonrenal sources. In either case, water and salt are lost, but the loss of sodium is greater than the loss of water.
1. Renal causes include the use of diuretics (particularly thiazide diuretics), osmotic diuresis, mineral corticoid deficiency/hypoaldosteronism, hypothyroidism, and, rarely, renal salt-wasting nephropathy, cerebral salt wasting, and certain types of renal tubular acidosis (RTA).
2. Nonrenal causes include fluid losses from the GI tract, skin (burns), cerebral salt wasting syndrome, and intravascular volume depletion via third spacing.
3. Renal and nonrenal causes can be distinguished by urine electrolytes. A urine sodium of >20 mEq/L suggests a renal source, while a urine sodium of <10 mEq/L suggests a nonrenal etiology.
4. The goal of therapy is to replace extracellular volume with isotonic sodium solutions and to allow for appropriate renal-free water excretion.
b. Hypervolemic hypotonic hyponatremia is associated with congestive heart failure (CHF), renal failure with nephritic/nephrotic syndrome, and cirrhosis.
1. Mechanism: In these disease processes, the effective arterial intravascular volume is low even if the total intravascular volume is normal or increased. The activation of the renin–angiotensin–aldosterone system and the sympathetic nervous system and the release of ADH lead to oliguria and salt retention. The result is expansion of ECF volume.
2. Manifestations include edema, elevated jugular venous pressures, pleural effusions, and ascites.
3. The goal of therapy is to control the primary disease process. Fluid and salt restriction and the use of proximal and loop diuretics may also be appropriate.
c. Isovolemic hypotonic hyponatremia
1. Causes: Syndrome of inappropriate ADH secretion (SIADH), psychogenic polydipsia, medications (e.g., oxytocin), physiologic nonosmotic stimulus for ADH release (e.g., nausea, anxiety, pain), hypothyroidism, and adrenal insufficiency.
2. Causes of SIADH include neurologic (head injury, subarachnoid hemorrhage, intracranial surgery), pulmonary (pneumonia, COPD, tuberculosis), malignancies, major surgeries (postoperative pain and nausea stimulate ADH secretion), and medications (desmopressin, cyclophosphamides, TCA, SSRI). It is important to contrast SIADH from cerebral salt wasting syndrome, a form of hypovolemic hyponatremia associated with head injury. The volume status is the only way to distinguish between SIADH and cerebral salt wasting in traumatic brain injury patients who are hyponatremic.
4. Therapy for hyponatremia
a. General guidelines: Most patients with hyponatremia have a chronic electrolyte abnormality and will be mostly asymptomatic. Thus, if these patients require treatment, it would be a slow correction through fluid restriction (euvolemic or hypervolemic etiology) or isotonic saline/oral salt tablets (hypovolemic or diuretic etiology). More aggressive therapy would be instituted in patients with symptomatic or severe hyponatremia, which would include hypertonic saline and potentially vasopressin receptor antagonists.
b. Treatment of underlying disease: If the patient is hyponatremic from adrenal insufficiency, administer glucocorticoids that will directly suppress the release of ADH. If the patient were hyponatremic due to reversible causes of SIADH (e.g., nausea, pain, anxiety), treat the underlying cause first. Iatrogenic/drug causes of hyponatremia should be approached by stopping the medication first.
c. Fluid restriction is an appropriate intervention for euvolemic and hypervolemic hyponatremia. The amount of fluid restriction is dependent on the level of urine output, but, in general, restriction to 50% to 60% of daily fluid requirements may be the treatment.
d. Urgent intervention is required in patients with symptomatic hyponatremia (nausea, vomiting, lethargy, altered level of consciousness, and seizures). The sodium deficit can be calculated as follows:
Sodium deficit = TBW × 140 − [Na]serum
The rate of correction of the serum sodium is important and must be tailored to the individual patient. Both delayed and rapid correction can be associated with neurologic injury. Safe correction can be achieved with hypertonic saline (3% NaCl) in euvolemic patients and with infusion of NS in hypovolemic patients. The rate of infusion should result in a correction of serum sodium of 1 to 2 mEq/L per hour for the first 24 hours or until serum sodium reaches a level of 120 mEq/L and then be reduced to a correction rate of 0.5 to 1 mEq/L/h.
e. The vaptans are active nonpeptide vasopressin receptor antagonists that are used in the treatment of euvolemic and hypervolemic hyponatremia including patients with chronic heart failure, cirrhosis, SIADH, or hyponatremia from other causes. Experience with these agents is limited, particularly in ICU patients.
C. Hypernatremia is defined by serum sodium of >145 mEq/L. Hypernatremia is also a description of total body sodium content relative to TBW and can exist in hypovolemic, euvolemic, and hypervolemic states. Hypernatremia could be due to water loss (more common) or increased salt intake without water. In all cases, the serum is hypertonic. Clinical manifestations of hypernatremia include tremulousness, irritability, spasticity, confusion, seizures, and coma. Symptoms are more likely to occur when the rate of change is rapid. When the change is gradual and chronic, cells in the CNS will increase the cellular osmolality, thereby preventing cellular water loss and dehydration. This process starts approximately 4 hours after the onset and stabilizes in 4 to 7 days. This change in CNS cellular osmolality is an important concept when considering therapy.
1. Hypovolemic hypernatremia
a. Caused by the loss of hypotonic fluids through extrarenal (e.g., skin or GI losses) or renal (e.g., osmotic diuresis and drug-induced) sources. There is loss of water and salt, with a greater proportion of water loss, which results in a decrease in the ECF volume and the effective arterial intravascular volume. Osmotic diuresis renally can be due to glucose, mannitol, or urea. GI losses include vomiting or osmotic diarrhea while skin losses include burns or perspiration.
b. It is recommended that isotonic saline be used for initial volume repletion, followed by hypotonic crystalloid solutions, such as 0.45% NS.
2. Euvolemic hypernatremia
a. Caused by the loss of free water through extrarenal (e.g., excessive insensible loss through skin or respiration) or renal (e.g., osmotic diuresis and drug-induced) sources.
b. Measuring urine osmolality (Uosm) is important. Extrarenal processes cause a high Uosm (>800 mOsm/kg H2O), whereas renal processes cause a low Uosm (approximately 100 mOsm/kg H2O).
c. In most cases of hypernatremia from free water loss, the intravascular and extracellular fluid volumes appear normal. Therapy involves replacement of free water.
d. Central and nephrogenic DI (see Chapter 26) are among the renal causes of euvolemic hypernatremia. Evaluation of urinary osmolality (usually urine osmolality is less than the plasma osmolality) and the response to ADH may help to determine the site of the lesion.
1. Central (neurogenic) DI can be caused by pituitary damage from tumor, trauma, surgery, granulomatous disease, and idiopathic causes. Central DI is treated with desmopressin (intranasal, 5–10 µg daily or twice daily).
2. Nephrogenic DI can be caused by severe hypokalemia with renal tubular injury, hypercalcemia, chronic renal failure, interstitial kidney disease, and drugs (e.g., lithium, amphotericin, demeclocycline). Therapy includes correcting the primary cause if feasible and possibly free water repletion.
3. Hypervolemic hypernatremia is caused by addition of excess sodium and usually results from the infusion or intake of solutions with high sodium concentration. The acute salt load leads to intracellular dehydration with ECF expansion, which can cause edema or CHF. The goal of therapy is to remove the excess sodium; this can be accomplished by using nonmedullary gradient disrupting diuretics (e.g., thiazides).
4. Free water deficit and correction of hypernatremia
Free water deficit = TBW * {1 − (140 / [Na])}
The correction of hypernatremia should occur at approximately 1 mEq/L/h. Approximately one-half of the calculated water deficit is administered during the first 24 hours and the rest over the following 1 to 2 days. Aggressive correction is dangerous, especially in chronic hypernatremia, where rapid correction can cause cerebral edema. Rapid correction is reasonable if the hypernatremia is acute (<12 hours). Neurologic status should be carefully monitored while correcting hypernatremia, and the rate of correction should be decreased if there is any change in neurologic function.
IV. POTASSIUM
A. In an average adult, the total body potassium is approximately 40 to 50 mEq/kg. Most of the potassium is in the ICF compartment. In general, intake and excretion of potassium are matched. The electrocardiogram (ECG) is useful in diagnosing true potassium imbalance because the level of polarization of an excitable cell and the ability for repolarization are determined by the extracellular and intracellular potassium concentrations.
B. Hypokalemia: serum potassium concentration of <3.5 mEq/L. A general rule is that a 1 mEq/L decrease in serum potassium represents approximately a total body potassium deficit of 200 to 350 mEq.
1. Causes of hypokalemia
a. Transcellular redistribution (increased entry into cells mostly through increased Na/K/ATPase pump)
1. Alkalemia (a 0.1–0.7 mEq/L change per 0.1 U change in pH).
2. Increased circulating catecholamines via β2 adrenergic receptors
3. Increased insulin.
b. Renal-associated causes
1. Without hypertension
a. With acidosis
i. Diabetic ketoacidosis (DKA) and renal tubular acidosis (RTA) types 1 and 2.
b. With alkalosis
i. Diuretics
ii. Vomiting
iii. Nasogastric suction (leading to hyperaldosteronism)
iv. Transport defects—thick ascending limb: Bartter’s syndrome; cortical collecting duct: Gitelman’s syndrome
2. With hypertension
a. Renal artery stenosis
b. Hyperaldosteronism due to tumor (Conn’s syndrome, adrenal adenoma)
c. Glucocorticoid-mediated hyperadrenalism
d. Pseudo-hyperadrenalism
i. Licorice ingestion
ii. Cushing’s syndrome
iii. Liddle’s syndrome
c. Hypomagnesemia
d. Acute leukemia
e. Excessive GI losses
f. Dietary
g. Lithium toxicity
h. Hypothermia
2. Manifestations of hypokalemia include myalgias, cramps, weakness, paralysis, urinary retention, ileus, and orthostatic hypotension. ECG manifestations, in order of progression of worsening hypokalemia, are decreased T-wave amplitude, prolonged QT interval, U wave, dragging of ST segment, and increased QRS duration. Arrhythmias are common, including atrial fibrillation, premature ventricular beats, supraventricular and junctional tachycardia, and Mobitz-I second-degree atrioventricular block (see Chapter 17).
3. Therapy: IV potassium replacement is appropriate in patients who have severe hypokalemia or who cannot take oral preparations. The rate of replacement should be governed by the clinical signs. The recommended maximal rate of infusion is 0.5 to 0.7 mEq/kg/h, with continuous ECG monitoring. Hypomagnesemia should be corrected prior to potassium repletion (see Section IV.C).
C. Hyperkalemia: serum potassium of >5.5 mEq/L.
1. Causes of hyperkalemia
a. Hemolysis of sample
b. Leukocytosis (white blood cell count >50,000/mm3)
c. Thrombocytosis (platelet count >1,000,000/mm3)
d. Transcellular redistribution:
1. Acidemia
2. Insulin deficiency, hyperglycemia, hyperosmolality
3. Drugs (digitalis, β-blockers, succinylcholine)
e. Malignant hyperthermia
f. Cell necrosis (rhabdomyolysis, hemolysis, burns)
g. Increased intake via replacement therapy and transfusions
h. Decreased renal potassium secretion
1. Renal failure
2. Hypoaldosteronism
3. Drugs: Heparin, angiotensin-converting enzyme inhibitors, and potassium-sparing diuretics
2. Manifestations of hyperkalemia include muscle weakness and cardiac conduction disturbances. ECG changes include atrial and ventricular ectopy (serum potassium of 6–7 mEq/L), shortened QT interval, and peaked T waves. Worsening hyperkalemia will lead to widening of the QRS and eventually ventricular fibrillation.
3. Therapy for hyperkalemia is emergent in the presence of ECG changes, particularly when serum potassium is >6.5 mEq/L. Continuous ECG monitoring is recommended. Therapy should begin in an ICU if the potassium levels show an increasing trend, even in the absence of ECG changes (see Chapter 23, Section VII.C).
V. ELECTROLYTE ABNORMALITIES: CALCIUM, PHOSPHORUS, AND MAGNESIUM
A. Calcium acts as a key signaling element for many cellular functions and is the most abundant electrolyte in the body. Normal values of total serum calcium range from 8.5 to 10.5 mg/dL (4.5–5.5 mEq/L). However, because calcium is bound to protein (approximately 40%), the appropriate range of total serum calcium that can provide for adequate ionized calcium is dependent on the total serum calcium and the amount of serum protein (particularly albumin). The ionized calcium provides a better functional assessment, with normal values ranging from 4 to 5 mg/dL (2.1–2.6 mEq/L, 1.05–1.3 mmol/L). Direct measurement of ionized calcium is commonly available and is superior to “corrected” calcium values based on albumin levels. Ionized calcium can be affected by the pH of the serum, with acidemia leading to higher ionized calcium and alkalemia to lower ionized calcium. Modulators of calcium homeostasis include PTH and 1,25-vitamin D, which increase calcium levels, and calcitonin, which decreases calcium levels.
1. Hypercalcemia (see Chapter 26): A total serum calcium >10.5 mg/dL or ionized calcium >5.0 mg/dL (2.6 mEq/L or 1.29 mmol/L)
a. Causes of hypercalcemia:
1. Primary hyperparathyroidism
2. Immobilization
3. Malignancy (bone destruction from metastases or hormone secretion)
4. Granulomatous diseases (tuberculosis, sarcoidosis), secondary to increased 1,25-vitamin D production by the granulomatous tissue
5. Thyrotoxicosis
6. Primary bone reabsorption abnormalities (Paget’s disease)
7. Adrenal insufficiency
8. Pheochromocytoma
9. Milk-alkali syndrome: High intake of calcium (>5 g/d)
10. Drugs (thiazides, vitamin D, lithium, estrogens)
b. Diagnosis
1. PTH levels: low in malignancy-associated hypercalcemia, high in primary, secondary, and tertiary hyperparathyroidism
2. 1,25-Vitamin D levels: elevated in granulomatous disease
3. PTH-related protein: elevated in malignancy-associated hypercalcemia (breast, lung, thyroid, renal cells)
4. Protein electrophoresis: monoclonal band associated with myeloma
5. Thyroid-stimulating hormone (TSH)
6. Chest radiographs: evaluate for malignancy and granulomatous disease
c. Manifestations: Hypercalcemia will affect multiple organ systems, including renal, GI, musculoskeletal, neurologic, and cardiovascular. Polyuria, nephrolithiasis, and nephrogenic DI are the renal effects. GI manifestations include nausea/vomiting, constipation, and pancreatitis. The patient will also exhibit muscle weakness, lethargy, and possibly coma. EKG abnormalities will include bradycardia, shortened QT interval, increased PR and QRS intervals, and atrioventricular block.
d. Treatment is described in detail in Chapter 26. Here we summarize the main considerations. Treatment should be initiated if neurologic symptoms are present, total serum calcium is >12 to 13 mg/dL, or calcium/phosphate product is >75.
1. Immediate hydration with NS to restore volume status and decrease serum calcium concentration by dilution
2. After establishing euvolemia, a loop diuretic can be added to NS with the goal of generating a urine output of 3 to 5 mL/kg/h.
3. Other electrolytes should be repleted.
4. Hemodialysis, if the above therapy is ineffective
5. The use of pamidronate, calcitonin, and glucocorticoids is described in detail in Chapter 26. Calcium-channel blockers can also be used to treat the cardiotoxic effects of hypercalcemia.
2. Hypocalcemia (see Chapter 26): an ionized calcium of <4 mg/dL
a. Causes of hypocalcemia:
1. Sequestration of calcium can be caused by hyperphosphatemia (from renal failure), pancreatitis, intravascular citrate (from packed red blood cells), and alkalemia.
2. PTH deficiency: PTH deficiency can be caused by surgical excision of parathyroid gland, autoimmune parathyroid disease, amyloid infiltration of parathyroid gland, severe hypermagnesemia, hypomagnesemia, HIV infection, and hemochromatosis.
3. PTH resistance: PTH resistance is due to congenital abnormality or secondary to hypomagnesemia.
4. Vitamin D deficiency is caused by malabsorption, poor nutritional intake, liver disease, anticonvulsants (phenytoin), inadequate sunlight, and renal failure.
5. Inappropriate calcium deposition can be due to formation of complex with phosphorus in hyperphosphatemic states (rhabdomyolysis), acute pancreatitis, and postparathyroidectomy.
6. Sepsis and toxic shock syndrome
b. Manifestations of hypocalcemia include generalized excitable membrane irritability leading to paresthesias and progressing to tetany and seizures. The classic physical examination findings include Trousseau’s sign (spasm of the upper extremity muscles that causes flexion of the wrist and thumb with the extension of the fingers and can be elicited by occluding the circulation to the arm) and Chvostek’s sign (contraction of ipsilateral facial muscles elicited by tapping over the facial nerve at the jaw). ECG changes include prolonged QT and heart block. Respiratory manifestations will include apnea, bronchospasm, and laryngeal spasm (typically seen after thyroidectomy or parathyroidectomy surgery). Anxiety and depression may also be noted as some neurologic indications of hypocalcemia.
c. Diagnosis
1. Confirm true hypocalcemia by checking ionized calcium and pH.
2. Rule out hypomagnesemia.
3. Check PTH level; if low or normal, hypoparathyroidism may be involved; if high, check for phosphorus level. A low phosphorus level is indicative of pancreatitis or vitamin D deficiency, while a high phosphorus level suggests rhabdomyolysis and renal failure.
d. Therapy of hypocalcemia: Infusion of calcium at 4 mg/kg of elemental calcium with either 10% calcium gluconate (93 mg of calcium/10 mL) or 10% calcium chloride (272 mg of calcium/10 mL). A bolus should be followed by an infusion because the bolus will increase the ionized form of calcium for 1 to 2 hours. To avoid precipitation of calcium salts, IV calcium solutions should not be mixed with IV bicarbonate solutions. Calcium chloride is caustic to peripheral veins and should be given via central venous access if possible. Suspected vitamin D or PTH deficiency is treated with calcitriol (0.25 µg, up to 1.5 µg PO once a day). Oral calcium repletion with at least 1 g of elemental calcium a day should be given together with vitamin D therapy.
B. Phosphorus exists mainly as a free ion in the body. Approximately 0.8 g to 1 g of phosphorus is excreted in the urine per day. Phosphorus excretion is affected by PTH (which inhibits proximal and distal nephron phosphorus reabsorption), vitamin D, high dietary phosphorus intake, cortisol, and growth hormone.
1. Hypophosphatemia occurs in 10% to 15% of hospitalized patients.
a. Causes
1. GI: malnutrition, malabsorption, vitamin D deficiency, diarrhea, and use of aluminum-containing antacids
2. Renal losses: primary hyperparathyroidism, renal transplantation, ECF expansion, diuretics (acetazolamide), Fanconi’s syndrome, post–obstructive and post–acute tubular necrosis (ATN), glycosuria, DKA
3. Redistribution: alkalosis, postalcohol withdrawal, parenteral hyperalimentation, burns, and continuous venovenous hemofiltration
b. Clinical symptoms usually occur when phosphorus is <1.0 mg/dL.
1. Neurologic: metabolic encephalopathy
2. Muscular: myopathy, respiratory failure, cardiomyopathy
3. Hematologic: hemolysis, white blood cell dysfunction
c. Diagnosis
1. Urinary phosphorus <100 mg/d implies GI losses.
2. Urinary phosphorus >100 mg/d suggests renal wasting.
3. Elevated serum calcium suggests hyperparathyroidism.
4. Elevated PTH suggests primary or secondary hyperparathyroidism or vitamin D–resistant rickets.
d. Treatment
1. Increase oral intake to 1,000 mg/d.
2. Elemental phosphorus, 450 mg per 1,000 kcal of hyperalimentation
3. Dose of IV phosphorus should not exceed 2 mg/kg (0.15 mmol/kg) of elemental phosphorus.
2. Hyperphosphatemia
a. Causes
1. Renal: decreased glomerular filtration rate (GFR), increased tubular reabsorption, hypoparathyroidism, pseudohypoparathyroidism, acromegaly, thyrotoxicosis
2. Endogenous: tumor lysis, rhabdomyolysis
3. Exogenous: vitamin D administration, phosphate enemas
b. Clinical symptoms are related to hypocalcemia due to calcium phosphate deposition and decreased renal production of 1,25-vitamin D.
c. Treatment
1. Phosphate binders to reduce GI absorption
2. Volume expansion and dextrose 10% in water with insulin may reduce acutely elevated phosphorus levels.
3. Hemodialysis and peritoneal dialysis
C. Magnesium: Serum magnesium is maintained between 1.8 and 2.3 mg/dL (1.7–2.1 mEq/L); 15% is protein bound.
1. Hypermagnesemia is rare in patients with normal renal function. It is defined as >2.5 mg/dL.
a. Causes
1. Acute and chronic renal failure
2. Magnesium administration for toxemia of pregnancy, magnesium-containing antacids and laxatives
3. Hypothyroidism
4. Lithium toxicity
b. Signs and symptoms
1. Cardiac dysrhythmias
2. Decreased neuromuscular transmission
3. CNS dysfunction: confusion, lethargy
4. Hypotension
5. Respiratory depression
c. Treatment
1. IV calcium
2. Hemodialysis to remove magnesium in renal failure
2. Hypomagnesemia is defined as serum magnesium <1.8 mg/dL.
a. Causes
1. GI
a. Decreased intake (chronic alcoholism)
b. Starvation
c. Magnesium-free enteral feedings
d. Decreased GI intake due to nasogastric suction and malabsorption
2. Renal losses
a. Diuretic therapy
b. Postobstructive diuresis
c. Recovery (polyuric phase) from ATN
d. DKA versus hypercalcemia
e. Primary hyperaldosteronism
f. Barter’s syndrome
g. Aminoglycoside, cisplatin, and cyclosporine nephrotoxicity
b. Manifestations
1. Hypokalemia and hypocalcemia; hypokalemia is the result of excess urine losses, which can only be corrected with magnesium repletion.
2. ECG changes mimic hypokalemia.
3. Digoxin toxicity is magnified by hypomagnesemia.
4. Neuromuscular fasciculations with Chvostek’s and Trousseau’s signs may be present.
c. Treatment for hypomagnesemia should be initiated when ECG changes and/or signs of tetany are present.
1. IV: with MgSO4, 6 g in 1 L of 5% dextrose in water over 6 hours.
2. Oral: with magnesium oxide, 250 to 500 mg four times a day.
VI. ACID–BASE PHYSIOLOGY: Acid–base homeostasis is essential for maintaining life. Significant acid–base derangements have major physiologic consequences that can be life threatening. However, it is the primary cause of the derangement that largely determines prognosis. Patients in the ICU often have important acid–base disturbances that require prompt diagnosis and treatment of the underlying condition and occasionally require symptomatic treatment of the acid–base disturbance itself. There are three major methods for quantifying acid–base disorders: the physiological approach, the base-excess approach, and the physicochemical (or Stewart) approach. In this section, we present a systematic approach to acid–base disorders relying primarily on the physiological approach.
A. Physiological Approach to Acid–Base Disturbances: The physiological approach uses the carbonic acid–bicarbonate system:
H+ + HCO−3 ↔ H2CO3 ↔ H2O + CO2
A primary change in PaCO2 causes a secondary (adaptive) response in the HCO3 concentration and vice versa. Since the concentration of hydrogen ions in plasma is very low (approximately 40 nEq/l), the pH (the negative logarithm of the hydrogen ion concentration) is used clinically. H2CO3 is in equilibrium with the dissolved CO2. The Henderson–Hasselbach equation describes the relationship between the pH, PaCO2, and HCO3:
pH = 6.1 + log {[HCO−3] / (0.03 × Paco2)}
Because pH = −log[H], the equation can be rearranged (this form is also known as the Henderson equation):
[H+] = 24 × (Paco2 / [HCO−3])
The above equation can be used to verify the reliability of a blood gas value, using the PaCO2 from the blood gas result, and a HCO3 from a simultaneously drawn metabolic panel (the HCO3 value reported in a blood gas is calculated, not measured). The [H+] is roughly estimated by adding or subtracting 10 nEq/L per change in 0.1 pH units from 40 nEq/L. The four primary acid–base disorders comprise two metabolic disorders (acidosis and alkalosis) and two respiratory disorders (acidosis and alkalosis).
B. A Systematic Approach to the Diagnosis of Acid–Base Disorders: All steps listed below are often not necessary. However, this approach enables an efficient and thorough evaluation for patients with complex acid–base abnormalities.
1. History and physical examination: A thorough clinical evaluation often provides important diagnostic clues. The medical history, including medications being taken, may predispose a patient toward specific acid–base disturbances. Vital signs, the presence of fever, neurological, pulmonary, and gastrointestinal signs and symptoms may indicate the severity of the disturbance as well as point to the type of acid–base disorder.
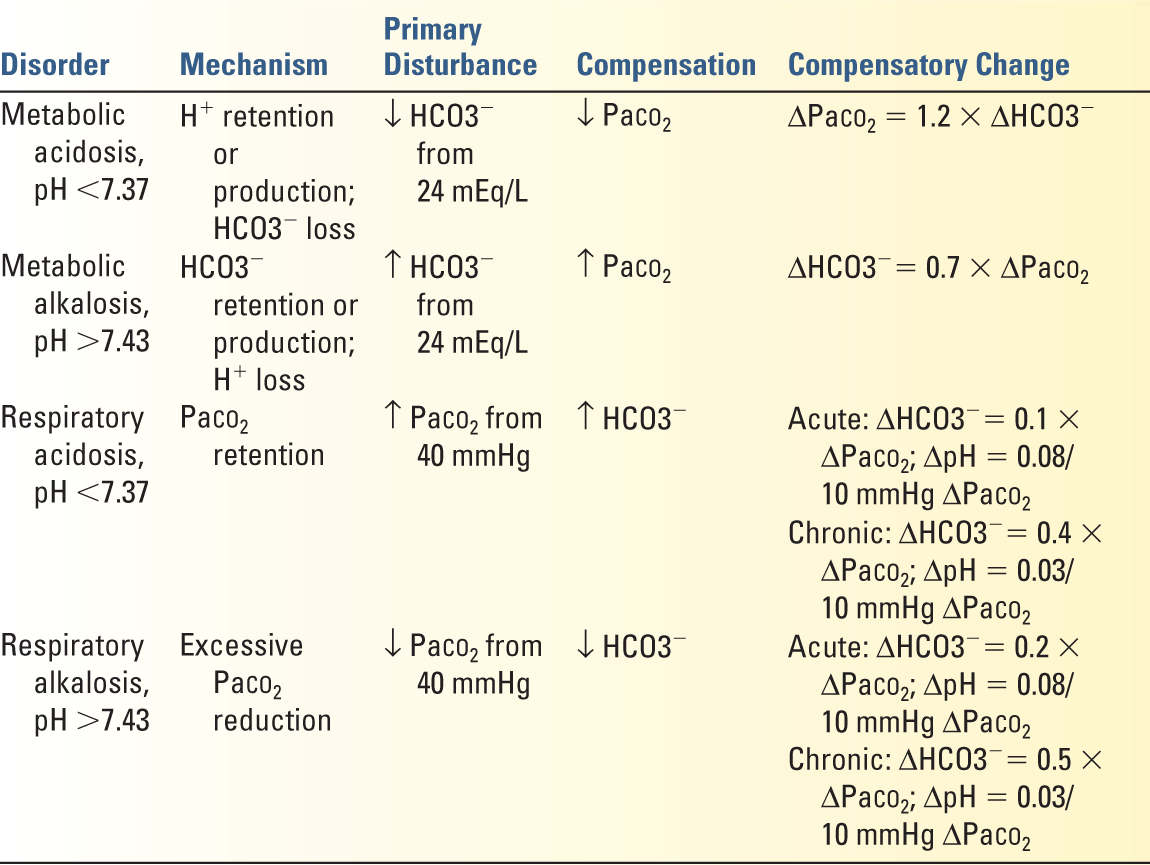
2. Determine the primary acid–base disorder: The pH defines whether the patient is acidemic (pH <7.38) or alkalemic (pH >7.42). The serum [HCO3−] (measured separately on a metabolic panel) determines whether there is a metabolic acidosis ([HCO3−] <22 mEq/L), or a metabolic alkalosis ([HCO3−] >26 mEq/L). The PaCO2 defines the presence of a respiratory acidosis (PaCO2 >42 mmHg) or respiratory alkalosis (PaCO2 <38 mmHg).
3. Define the secondary (or compensatory) response: The predicted PaCO2 for a given [HCO3−] or vice versa can be calculated (Table 8.5). The presence of a mixed acid–base disorder (i.e., two or even three coexisting acid–base disturbances) is suggested if the measured value is greater or less than predicted. For example, a patient with a pH of 7.16 with a [HCO3−] of 12 mEq/L has a primary metabolic acidosis. The predicted PaCO2 for this patient would be 26 mmHg. If the actual PaCO2 is 40 mmHg, the patient has a superimposed respiratory acidosis (even though the PaCO2 is “normal”).
4. Calculate the corrected anion gap (AGcorr): For a metabolic acidosis, the AG is calculated ([Na+] 2 [Cl−] − [HCO3−]). The normal value is typically between 5 to 12 mEq/L, depending on the laboratory. Since albumin accounts for up to 75% of the anion gap, the AG should always be corrected for hypoalbuminemia (common in critically ill patients) to give the corrected anion gap (AGcorr = AG + 2.5 × (4.0 − Pt Albumin (in g/dL)).






Full access? Get Clinical Tree


