171 Ethanol, Methanol, and Ethylene Glycol
 Ethanol Intoxication
Ethanol Intoxication
Ethanol is rapidly absorbed by the gastrointestinal tract and distributed throughout body water.1 The blood ethanol concentration (in mg/dL) resulting from a one-time dose can be estimated from the volume (in mL) of ingested alcoholic beverage, the fractional concentration of ethanol (by volume) in the beverage, and body weight (in kg), by the following equation:
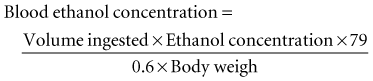
Metabolism
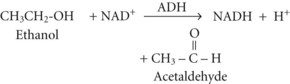
Between 2% and 10% of ingested ethanol is excreted intact by the kidneys and lungs, but the major fraction is metabolized by hepatic alcohol dehydrogenase (ADH) to acetaldehyde by the following reaction2:
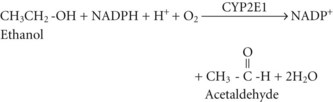

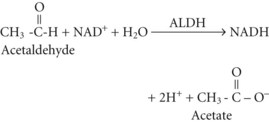
Acetate can then enter the tricarboxylic acid cycle and ultimately be metabolized to carbon dioxide (CO2) and water. Polymorphisms in the dehydrogenase enzymes can result in increased production rates or diminished metabolic clearance of acetaldehyde. As a consequence, some individuals experience marked vasodilation, facial flushing, tachycardia, and other unpleasant symptoms after ethanol consumption because of the effects of excessive acetaldehyde accumulation. Alleles leading to this reaction are particularly prevalent in persons of Chinese or Japanese descent but are uncommon in Caucasians.2
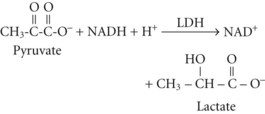
The resulting increase in blood lactate level is usually small, however, and the presence of lactic acidosis should prompt consideration of an alternative cause such as circulatory shock or seizures.3
Clinical Manifestations
Acute intoxication can induce cardiac dysrhythmias, particularly atrial fibrillation. As denoted by the descriptive sobriquet, “holiday heart syndrome,” this phenomenon frequently occurs during an alcoholic binge. A variety of neurologic abnormalities are associated with chronic alcoholism, including Wernicke-Korsakoff syndrome, chronic cerebellar ataxia, Marchiafava-Bignami syndrome, and central pontine myelinolysis.4 Wernicke encephalopathy can manifest as lethargy, confusion, truncal ataxia, nystagmus, and ophthalmoplegia, whereas Korsakoff dementia manifests as retentive memory impairment, confabulation, and learning deficits.5
Acutely, ethanol has well-known, dose-dependent inebriating and sedating effects (Table 171-1), although remarkable variability in this relationship is observed in some individuals.4 These central nervous system (CNS) effects appear to be at least partly caused by interference with N-methyl-D-aspartate receptor and perhaps γ-aminobutyric acid receptor function.4,6,7 The cognitive, behavioral, perceptual, and psychomotor effects of ethanol intoxication play a causative role in a substantial proportion of deaths and injuries involving motor vehicle–related trauma, accidental drownings, residential fires, homicides, and suicides. The legal driving threshold for blood ethanol concentration is 80 mg/dL in the United States for operators aged 21 years or older. Tachycardia, mydriasis, diaphoresis, hypotension, and hypothermia can occur in cases of marked intoxication. Blood ethanol concentrations of approximately 350 mg/dL have been associated with fatal outcomes, although many patients have survived much higher levels, including one subject who reportedly survived a level of 1500 mg/dL.8
TABLE 171-1 Relationship Between Blood Ethanol Concentration and Clinical Manifestations*
| Blood Ethanol Concentration (mg/dL) | Clinical Manifestations |
|---|---|
| <30 | Little demonstrable effect |
| 30-50 | Mild euphoria, minimal central nervous system effects, subjective sensation of cutaneous warmth |
| 50-80 | Relaxation, jocularity, gregariousness, cutaneous flushing, prolongation of reaction time |
| 80-100 | Statutory intoxication in many jurisdictions |
| 100-200 | Loquacity, animation, exuberance, exaggerated emotional responses, uninhibited behavior, impaired judgment |
| 200-300 | Sedation interrupted by periods of boisterous or antisocial behavior, nausea, emesis, dysarthria, horizontal nystagmus, impaired visual pursuit, diplopia, ataxia |
| 300-400 | Unstable station and gait, incoherent speech, somnolence, impairment of protective airway reflexes, incontinence, obtundation, stupor |
| >400 | Coma, loss of protective reflexes, respiratory depression, death |
* This information serves only as an imperfect guide, because considerable variability and overlap is possible, and individuals with chronic heavy ethanol exposure often develop learned tolerance.
Laboratory Manifestations
Blood ethanol concentration correlates at least approximately with the manifestations of intoxication (see Table 171-1). In chronic alcoholic subjects, a blood ethanol concentration below 250 mg/dL is an unlikely explanation for alterations in consciousness and should prompt a search for an alternative cause.8 Numerous other blood test abnormalities can be seen in intoxicated subjects, particularly in patients with chronic ethanol abuse: hyponatremia, hypokalemia, hypomagnesemia, hypophosphatemia, hypoglycemia, thrombocytopenia, and coagulopathy. Elevated activities of various circulating enzymes including amylase, lipase, creatine phosphokinase, transaminases, and γ-glutamyl transpeptidase, can occur as a reflection of alcohol-induced pancreatitis, rhabdomyolysis, hepatitis, or cirrhosis. The latter can also result in hyperbilirubinemia and hypoalbuminemia.
Treatment
In the absence of associated illness or injury (Table 171-2), mild to moderate intoxication requires no special treatment other than abstinence and a period of observation. Regardless of the degree of intoxication, withdrawal precautions are recommended for chronic imbibers, particularly those with a history of heavy chronic use or alcohol withdrawal manifestations. The treatment of severe ethanol intoxication is largely supportive. As with any patient who presents to the hospital in an unconscious state, initial empirical treatment should include IV thiamine, dextrose, and naloxone, once adequate airway, ventilation, and perfusion are ensured. Gastric lavage and activated charcoal administration are of dubious value for hastening removal of ethanol from the body.9–12
TABLE 171-2 Concomitant or Complicating Disorders Associated with Alcohol Intoxication or Withdrawal
| Alcoholic hepatitis | Hypoglycemia |
| Aspiration pneumonitis | Hypothermia |
| Circulatory shock (due to dehydration or hemorrhage) | Infections (e.g., pneumonia, meningitis) |
| Cirrhosis | Intracranial hemorrhage (e.g., subdural hematoma) |
| Coagulopathy | Pancreatitis |
| Dehydration | Peripheral neuropathy |
| Drug overdose or other toxic ingestion | Psychosis |
| Electrolyte derangements | Rhabdomyolysis |
| Gastrointestinal hemorrhage (due to gastritis, peptic ulcer disease, esophageal varices, hemorrhoids, or Mallory-Weiss tear) | Seizures |
| Sepsis | |
| Head injury | Thrombocytopenia |
| Heat stroke | Vitamin deficiency (folate, thiamine, other B vitamins) |
| Hepatic encephalopathy | Wernicke-Korsakoff syndrome |
A thorough evaluation for common associated illnesses and injuries should include physical and laboratory examinations for evidence of head, neck, and somatic trauma, rhabdomyolysis, pancreatitis, hepatic dysfunction, coagulopathy, blood dyscrasias, and fluid and electrolyte derangements. Accordingly, routine laboratory testing should include a complete blood count, prothrombin and partial thromboplastin times, serum assays for electrolytes (including sodium, potassium, chloride, total CO2 content, magnesium, and phosphorus), glucose, liver and kidney function tests, and amylase, lipase, transaminases, and creatine phosphokinase activities. Screening for alternative or concomitant intoxications or overdoses is occasionally fruitful.13 Identification of metabolic acidosis should prompt investigation for alcoholic ketoacidosis, lactic acidosis, renal failure, and relevant toxic ingestions, particularly methanol and ethylene glycol. Microbiological cultures are indicated if there are signs of serious infection.
Intravenous thiamine and a multivitamin preparation containing folate are routinely administered to hospitalized patients with alcohol intoxication or withdrawal. Parenteral thiamine (50 or 100 mg) is given during the initial phase of management, regardless of the level of sensorium, to prevent or treat Wernicke-Korsakoff syndrome.5
Oxygenation may be assessed either by pulse oximetry or by arterial blood gas analysis, and supplemental oxygen should be provided as necessary. Administration of vitamin K, fresh frozen plasma, or platelet transfusions may be necessary if there is gastrointestinal or other hemorrhage and coagulopathy or severe thrombocytopenia. The level of consciousness should be monitored periodically. Hemodialysis has been employed and is effective at removing ethanol from the body, but in general, this modality poses greater risks than simply providing supportive care and allowing physiologic ethanol elimination. Its use might be warranted in rare cases of profound life-threatening ethanol intoxication, or if there are other reasons for dialysis.14,15
 Alcoholic Ketoacidosis
Alcoholic Ketoacidosis
Metabolism
Although the precise metabolic mechanisms that lead to the development of AKA are incompletely understood, several mechanisms appear to be operative. Abnormal insulin and counterregulatory hormone levels occur,16 but the disorder is distinct from simple starvation and diabetes mellitus. Ethanol results in inhibition of gluconeogenesis and depletion of glycogen stores, leading to low glucose availability, particularly when coupled with fasting. Hypoglycemia causes release of epinephrine, cortisol, and growth hormone, as well as decreased insulin production; these are all factors that favor ketone synthesis. Ethanol metabolism results in a surfeit of acetate and NADH, which promotes lactate and ketone production. Marked ketonemia results in acidosis and ketonuria. The latter causes osmotic diuresis, intravascular volume depletion, and electrolyte losses. Thus, starvation, dehydration, excessive acetate production, an altered redox state, hormonal imbalances, and perhaps genetic predisposition are all potentially involved.17
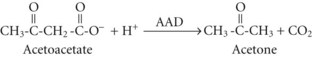
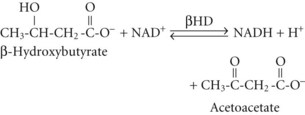
In both AKA and diabetic ketoacidosis (DKA), β-hydroxybutyrate is quantitatively the more important molecule. However, the ratio of β-hydroxybutyrate to acetoacetate tends to be higher in AKA (typically 5 : 1 but sometimes exceeding 10 : 1),18 compared with DKA (typically 3 : 1).
Clinical Manifestations
AKA characteristically develops 24 to 72 hours after an alcoholic debauch as the blood ethanol concentration is declining, during which time the subject ceases ethanol consumption and has little or no caloric intake. Gastrointestinal symptoms predominate and include anorexia, nausea, epigastric pain, and vomiting.19,20 The subject usually has a temporary aversion to food and alcoholic beverages and complains of malaise. On physical examination, there is a clear sensorium in most cases. The odor of acetone may be detectable on the subject’s breath. Tachypnea or Kussmaul respirations may be evident if there is marked acidemia. Tachycardia and other signs of volume depletion may be apparent. In some cases, manifestations of underlying cirrhosis (e.g., jaundice, ascites, ecchymoses, hemorrhoids) or other disorders commonly associated with chronic alcohol abuse (see Table 171-2) may be present.
Laboratory Manifestations
The key laboratory findings in AKA are metabolic acidosis, ketonemia, and ketonuria in the presence of a normal, low, or mildly elevated blood glucose concentration. Ethanol may be detectable in the blood, but it is not a requirement for the diagnosis and is frequently not detectable by the time the patient presents to the hospital. If the acidosis is clinically significant, elevation of the serum anion gap is expected. Other causes of metabolic acidosis must be excluded. Simple starvation can cause mild ketoacidosis, but with simple starvation the serum total CO2 content or bicarbonate concentration generally remains above 18 mmol/L. DKA and renal failure are readily excluded by routine blood glucose and creatinine measurements. Lactic acidosis may be suggested by the associated clinical setting (e.g., seizures, hypotension), but it should be excluded by direct assay. Mild degrees of hyperlactatemia can occur in AKA, but concentrations greater than 3 mmol/L should prompt consideration of occult hypoperfusion, seizures, or another cause. Occult toxic ingestions also require exclusion, particularly ingestions of methanol, ethylene glycol, and salicylate intoxication.15,21–24 Ingestion of exogenous acetone or isopropanol can cause marked ketosis due to acetonemia, but in isolation these intoxications are not associated with anion gap elevation or metabolic acidosis unless the poisoning is severe enough to cause seizures or circulatory shock, thereby resulting in lactic acidosis.
Because vomiting and dehydration are frequent manifestations in AKA, metabolic alkalosis can complicate the acid-base derangement. The combination of metabolic acidosis (from ketoacidosis) and metabolic alkalosis (from vomiting and volume contraction) can result in arterial pH and blood gas values that underestimate the severity of one or both of these metabolic disturbances. For example, mild metabolic alkalosis can be obscured by the presence of moderate or severe metabolic acidosis. Rarely, both metabolic processes are present and of approximately equal severity. In this situation, blood pH and bicarbonate concentration can be within normal limits despite the acid-base disturbances.23 Or, the metabolic alkalosis can predominate and obscure the acidosis. The serum anion gap can aid in detecting these situations. An abnormally high anion gap suggests metabolic acidosis even if no acid-base disorder is evident by arterial blood gas analysis. In the face of a wide serum anion gap, the quotient of the delta anion gap (i.e., the subject’s anion gap minus the average normal anion gap) divided by the delta bicarbonate (i.e., the average normal bicarbonate concentration minus the subject’s blood bicarbonate concentration) should equal unity in organic metabolic acidoses if there is no metabolic alkalosis.25 A quotient well above unity (e.g., >1.2) is evidence of concomitant metabolic alkalosis.
Treatment
Alternative explanations for the metabolic acidosis should be promptly excluded.24 As in acute alcohol intoxication, the initial assessment should focus on identifying relevant alternative, underlying, or complicating illnesses or injuries that may require specific urgent therapy. Although patients with AKA sometimes have severe metabolic acidemia, the acid-base disturbance usually responds rapidly to IV hydration and ample dextrose administration.17 Rapid infusion of 50 mL of 50% dextrose is indicated if hypoglycemia is identified. Five percent dextrose in normal saline is infused IV, at a high rate initially, to correct any hypovolemia or hypoglycemia and provide substrate for metabolic correction of the ketoacidosis. Thereafter, dextrose-containing normal or half-normal saline can be substituted at a high maintenance infusion rate, titrated to ongoing fluid losses. Ample dextrose administration is key to reversing the metabolic acidosis. The blood glucose concentration should be monitored frequently to allow detection of recurrent hypoglycemia or any intolerance to the provided glucose load.
 Ethanol Withdrawal
Ethanol Withdrawal
Ethanol withdrawal is common among hospitalized patients, either as a primary reason for admission or as a development during hospitalization for some other illness or injury. It is a potentially fatal syndrome that occurs after abrupt discontinuation of ethanol in individuals who regularly consume ethanol-containing beverages. Although in most cases it occurs after complete abstinence, it can also occur in the face of ongoing ethanol consumption if the level of ethanol intake is substantially decreased. The pathophysiology is incompletely understood but probably involves changes in neurotransmitter levels and alterations in neurotransmitter receptor function, as well as elevated circulating catecholamine levels.6,7,26,27 A number of disorders should be of particular consideration in the differential diagnosis of alcohol withdrawal (see Table 171-2). The mortality rate associated with advanced stages of alcohol withdrawal can exceed 15%.28,29
Clinical Manifestations
The syndrome is traditionally classified into four stages, although the stages do not always follow the indicated sequence, and not every patient develops every stage.29 The time of development of each stage is also quite variable, and overlaps can occur. A typical temporal sequence is described.
The first stage occurs 6 to 24 hours or more after the last drink or after a somewhat longer period of markedly decreased ethanol intake. Manifestations include anxiety, restlessness, decreased attention, tremulousness, insomnia, and craving for alcoholic beverages. Stage 2, which occurs about 24 hours after the onset of abstinence, is characterized by hallucinations, misperceptions, irritability, and vivid dreams.30 Hallucinations may be auditory, but more often they are visual or tactile. Formication, the delusional sensation of insects crawling on the skin, and vivid or threatening visual hallucinations are particularly common. During this stage, the patient may appear otherwise lucid or somewhat confused, hypervigilant, and easily startled or misled. In stage 3, which commonly occurs 7 to 48 hours after cessation of drinking, seizures occur, usually of the grand mal variety.4 The seizures classically manifest as a cluster of brief tonic-clonic convulsions, at one time referred to as “rum fits.” They are more likely to occur in subjects with a history of repeated withdrawal episodes.32 A relatively lucid interval ranging from hours to 2 or 3 days is sometimes seen between stages 3 and 4. Stage 4 manifests 2 to 6 days or more after initiation of abstinence and consists of a global confusional state associated with signs of neuronal excitation and severe autonomic hyperactivity. Vernacular usage notwithstanding, the term delirium tremens specifically refers to stage 4 of withdrawal. Only a small minority of individuals with alcohol withdrawal develop delirium tremens. Tremors, hallucinations, and seizures are common during this stage. As is characteristic of delirium in general, the degree of confusion and disorientation can wax and wane. Hyperadrenergic manifestations may include diaphoresis, flushing, mydriasis, tachycardia, hypertension, and low-grade fever.4
Treatment
Initial steps in management include ensuring that a patent airway is present and that ventilation, oxygenation, and perfusion are adequate; establishing IV access; and excluding serious coexisting or complicating disorders. Subsequent treatment focuses mainly on judiciously titrated sedation and vigilant monitoring for progression of the syndrome or development of complications. All patients with alcohol withdrawal are given prophylactic multivitamin supplements including parenteral thiamine and folate, and fluid deficits and electrolyte deficiencies are corrected.33 Routine administration of magnesium sulfate in the absence of hypomagnesemia has not been shown to be beneficial.34,35 Prophylaxis against deep vein thrombosis is recommended.
The agent of choice is a benzodiazepine given orally in milder cases or IV in more severe withdrawal states.30,33,36–38 Limited evidence suggests that symptom-triggered dosing is superior to fixed-schedule benzodiazepine dosing.39 Individualized dosing requires the expert judgment of an experienced clinician, but practicality often necessitates substitution of protocol-driven dosing schemes. These typically use a quantitative assessment scale such as the Revised Clinical Institute Withdrawal Assessment Scale for Alcohol to score the degree of withdrawal manifestations.40,41 Lorazepam can be administered IV in incremental doses, starting with 1 or 2 mg, followed by intermittent (e.g., every 2-6 hours) IV dosing or a continuous IV infusion (e.g., initiated at 1 mg/h and titrated to effect).29,42 Alternatively, midazolam can be employed, beginning with 2 to 4 mg by IV injection, followed by 2 mg/h by continuous IV infusion, which may be titrated to effect. Diazepam is another option, given initially in titrated doses of 5 to 10 mg at intervals as frequent as every 10 minutes if necessary until a calm but awake level of consciousness is achieved. Subsequent dosing at 5 to 20 mg every 4 to 6 hours is typically required with this agent. Prolonged administration of diazepam can lead to prolonged duration of sedation due to accumulation of the parent drug and an active metabolite, both of which have long half-lives. This effect is less likely to occur with lorazepam.
Oral benzodiazepines have been employed commonly in mild cases of withdrawal that do not require IV sedation.30,31 These agents also can be used in more serious cases after the severe manifestations have abated and parenteral benzodiazepines are no longer required. Typical oral chlordiazepoxide dosage is 25 to 100 mg every 6 to 12 hours. Intramuscular administration is sometimes employed, but it entails a less predictable dose-response due to erratic absorption, and there is the potential for a depot effect.
Other sedative-hypnotic drugs can be effective but are not considered first-line therapeutic agents.33,36 Barbiturates have a long history of successful use. The most commonly used agent is phenobarbital, which can be difficult to titrate because of its long duration of action. The shorter-acting barbiturate, pentobarbital, also has been employed. Oral ethanol and, in the past, paraldehyde have been used but have been discouraged, in part because of the risks of aspiration and gastric irritation, but also because their use can be interpreted as reinforcing the acceptability of using alcoholic beverages, either in general or for treatment of withdrawal symptoms. The latter criticism has also been directed at the use of ethanol administered IV for this purpose. A randomized trial examining IV ethanol administration for alcohol withdrawal prophylaxis in trauma ICU patients found no advantage compared to benzodiazepine management.43 Propofol is effective, but it is not a first-line agent and is not recommended unless an endotracheal tube is in place and mechanical ventilation is used.29 Regardless of the specific sedative agent employed, appropriate dose titration is crucial. The goal is to ameliorate the manifestations of withdrawal without causing excessive sedation. Sedation should be titrated with the use of an objective sedation scale such as the Ramsay Sedation Scale,44 the Riker Sedation-Agitation Scale,45 or the Richmond Agitation-Sedation Scale.46 The goal should be to achieve a calm awake state or, if that is not feasible, a state of light somnolence from which the patient can easily be aroused and is able to respond verbally.
Clonidine may be administered if hyperautonomic symptoms are prominent.47–49 Typical oral dosing is 0.1 to 0.2 mg every 6 to 12 hours. β-Adrenergic receptor blockers are not recommended for routine use, but barring contraindications, they may be considered in selected cases as adjunctive agents for controlling severe hyperadrenergic manifestations. Haloperidol and other neuroleptic agents are not routinely used, because they can lower the threshold for seizures. In selected cases, haloperidol may be used in conjunction with benzodiazepines for marked agitation or hallucinations, but this agent or similar drugs should probably not be used as monotherapy.36
Seizure precautions should be instituted for all patients in withdrawal. Withdrawal seizures are managed primarily with benzodiazepines, which usually are effective at the doses used for sedation.42 In refractory cases, higher doses may be necessary but may necessitate endotracheal intubation and mechanical ventilation. Concomitant use of other anticonvulsants also can be considered. Barbiturates may be used for this purpose, but phenytoin is usually ineffective unless the seizures are due to a specific cause other than alcohol withdrawal, such as underlying epilepsy or a complicating acute disorder of the CNS (e.g., meningitis, head trauma).33,50,51 In such cases, phenytoin is usually the anticonvulsant of choice. A variety of other anticonvulsant and sedative drugs have been studied for potential use in treating alcohol withdrawal, including valproic acid, baclofen, γ-hydroxybutyrate, gabapentin, oxcarbazepine, and carbamazepine. However, data on safety and efficacy are limited, particularly for hospitalized patients and those with comorbid illness.52–59



Full access? Get Clinical Tree


