Critical Care Medicine
Adult Critical Care
Angiotensin-Converting Enzyme N-Terminal Inactivation Alleviates Bleomycin-Induced Lung Injury
Li P, Xiao HD, Xu J, et al (Cedars-Sinai Med Ctr, Los Angeles, CA; Emory Univ, Atlanta, GA; et al) Am J Pathol 177:1113-1121, 2010§
Evidence Ranking
• A
Expert Rating
• 1
Abstract
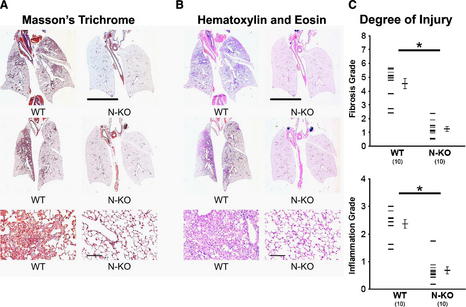
Bleomycin has potent anti-oncogenic properties for several neoplasms, but drug administration is limited by bleomycin-induced lung fibrosis. Inhibition of the renin-angiotensin system has been suggested to decrease bleomycin toxicity, but the efficacy of such strategies remains uncertain and somewhat contradictory. Our hypothesis is that, besides angiotensin II, other substrates of angiotensin-converting enzyme (ACE), such as the tetrapeptide N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP), play a significant role in controlling fibrosis. We studied bleomycin-induced lung injury in normotensive mice, termed N-KO and C-KO, which have point mutations inactivating either the N- or C-terminal catalytic sites of ACE, respectively. N-KO, but not C-KO mice, have a marked resistance to bleomycin lung injury as assessed by lung histology and hydroxyproline content. To determine the importance of the ACE N-terminal peptide substrate AcSDKP in the resistance to bleomycin injury, N-KO mice were treated with S-17092, a prolyl-oligopeptidase inhibitor that inhibits the formation of AcSDKP. In response to bleomycin injection, S-17092-treated N-KO mice developed lung fibrosis similar to wild-type mice. In contrast, the administration of AcSDKP to wild-type mice reduced lung fibrosis due to bleomycin administration. This study shows that the inactivation of the N-terminal catalytic site of ACE significantly reduced bleomycin-induced lung fibrosis and implicates AcSDKP in the mechanism of protection. These data suggest a possible means to increase tolerance to bleomycin and to treat fibrosing lung diseases (Figs 1, 2, and 8).
Related posts:
 of change in daily step count over five years with insulin sensitivity and adiposity: population based cohort study
of Cancer: The Next Generation
Beverages and Risk of Metabolic Syndrome and Type 2 Diabetes: A meta-analysis
time and haemodynamic response after thiopental vs. propofol in the elderly: a randomized trial
monitoring of neuromuscular block over the orbicularis oris muscle in anesthetized patients receiving vecuronium
Is Better Than General Anesthesia During Endovascular Acute Stroke Interventions
of change in daily step count over five years with insulin sensitivity and adiposity: population based cohort study
of Cancer: The Next Generation
Beverages and Risk of Metabolic Syndrome and Type 2 Diabetes: A meta-analysis
time and haemodynamic response after thiopental vs. propofol in the elderly: a randomized trial
monitoring of neuromuscular block over the orbicularis oris muscle in anesthetized patients receiving vecuronium
Is Better Than General Anesthesia During Endovascular Acute Stroke Interventions

Full access? Get Clinical Tree


