MECHANISMS OF CRITICAL ILLNESS–ASSOCIATED DYSREGULATION OF THE ENDOCRINE SYSTEM
The Stressors
Critically ill patients are unexpectedly faced with a life-threatening situation and a hostile environment (the intensive care unit). Physical stressors may be disease related (e.g., infection, burns, trauma) or interventions related (e.g., surgery, invasive diagnostic or therapeutic procedures, drugs). Their unpredictable nature, duration, and intensity render host response more complex and less likely to be appropriate.
Mechanisms of Neuroendocrine Activation in Acute Inflammation
On the one hand, acute inflammatory response to insults, for example, lipopolysaccharide (LPS) includes the release of a broad variety of mediators, for example, tumor necrosis factor alpha (TNF-α), interleukin (IL)-1, IL-6, IL-8, nitric oxide (NO), macrophage migration–inhibiting factors (MIF), and high mobility group box (HMGB)-1 (3,4) (Fig. 136.4). These mediators enter into hypophyseal portal capillaries within the median eminence, where they can diffuse into the pituitary-free of blood–brain barrier (5) and then to the hypothalamus. They also diffuse to the brain via other areas lacking a blood–brain barrier (organum vasculosum lamina terminalis, subfornical organ, subcommissural organ, area postrema, pineal gland, and probably the plexus choroids), or by crossing the barrier using specific transport systems (6). Microglial cells—brain’s resident macrophages—can produce cytokines, for example, IL-1, IL-2, and IL-6 (7). Following LPS (8) or sepsis (9), both pituitary and hypothalamic tissues expressed various cytokines via an inducible NO synthase (iNOS)–mediated pathway (10,11). Sepsis was associated with a loss in CRH and AVP regulation of ACTH synthesis, and iNOS-related decrease in ACTH synthesis (12). The NO regulatory effects are mediated by combined activation of guanylate cyclase, cyclooxygenase, and lipoxygenase (13). Cytokines can directly stimulate ACTH synthesis and release (14). Brain also expressed anti-inflammatory mediators, for example, IL-10, IL-13, and IL-1 receptor antagonists in pituitary and pineal glands (15). On the other hand, terminal nerve endings have receptors for numerous danger-associated molecular pattern receptors and may sense inflammatory threats at tissue levels, and subsequently signal to the noradrenergic system and the hypothalamus (1). Following LPS, vagal afferent fibers activated neurons of locus coeruleus which have projections synapsing on cholinergic interneurons in the parvocellular nucleus (16). Then, CRH was released upon acetylcholine stimulation of muscarinic receptors (17), an effect prevented by nonspecific NO antagonists (16). Finally, endogenous gas such as carbon monoxide (18) and hydrogen sulfide (19) may variably down- or upregulate hypothalamic and pituitary cells.
Variations in Cytokine-Induced Activation of the Hypothalamic–Pituitary Axis
IL-1 was the first cytokine shown to strongly and sustainably stimulate CRH release (20), whereas IL-6 and TNF induced weak and transient hypothalamic responses, and IL-2 and interferon alpha had no effect (21). Endocrine effects of cytokines may be dependent on route of administration (22). Cyclooxygenase inhibitors fully prevented hypothalamic response to intravenous IL-1 challenge, but only prevented early response to an intraperitoneal challenge, suggesting a biphasic response to IL-1 (20,23). Cyclooxygenase inhibitors also attenuated hypothalamic response to TNF but not to IL-6 (21). The noradrenergic system, mainly substance P, neuropeptides Y, or galanin, modulated hypothalamic response to IL-1 whereas the serotoninergic system had no effect (24).
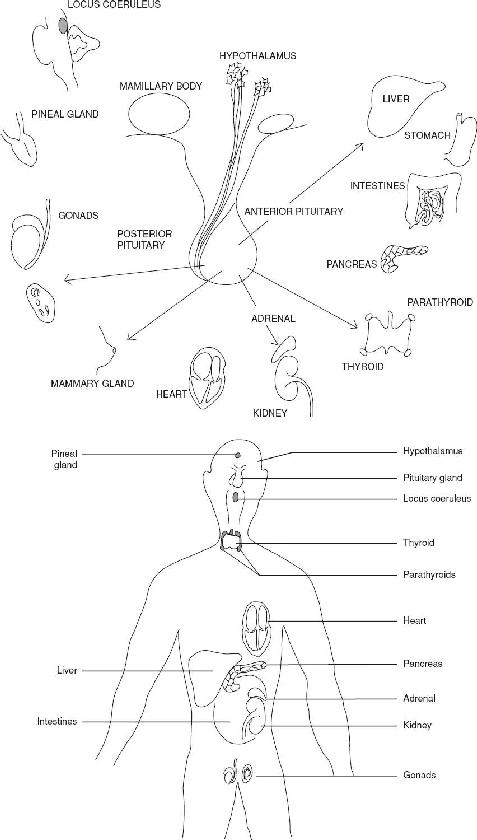
FIGURE 136.1 The endocrine organs are distributed throughout the body. There is an electrochemical connection from the hypothalamus to all organs, controlling body metabolism, growth and development, and reproduction.
| TABLE 136.2 List of Hormones |
 |
 |
 |

FIGURE 136.2 The hypothalamus lies directly above the pituitary gland. The hypothalamic neurohypophysial tract defines the neuronal system terminating in the posterior pituitary and is best known for its secretion of vasopressin and oxytocin into the peripheral circulation. The pars distalis of the anterior pituitary is supplied by venous blood delivered through the long portal veins that descend along the ventral surface of the pituitary stalk and interconnect capillary beds in the pars distalis with specialized capillary beds of the portal capillary system in the base of the hypothalamus called the median eminence. Venous drainage from the anterior pituitary to the systemic circulation is through adenohypophyseal veins.
DIAGNOSIS
Hormonal Patterns during Critical Illness
Infection, LPS challenge, major surgery, trauma, or burns elicit very similar patterns of pituitary hormone secretion. Plasma ACTH and prolactin increase within a few minutes following the insult and are associated with a rapid inhibition of LH and TSH but not FSH. Growth hormone secretion is also stimulated in humans but inhibited in rats (14).
Hypothalamic–Pituitary–Adrenal Axis
Acute stress increases the amplitude and frequency of ACTH and cortisol pulses and circadian rhythm is lost (1). To that purpose, additional secretagogues of CRH, vasopressin, and ACTH are recruited mainly magnocellular vasopressin and angiotensin II. In addition, catecholamines, neuropeptides Y, and to a lesser extent CRH released from the adrenal medulla, as well as direct autonomic neural input to the adrenal cortex, stimulate glucocorticoid secretion. The common feature is characterized by high circulating levels of ACTH and cortisol, which remain in plateau as long as the stressful condition is maintained. However, in critical illness, circulating levels of cortisol reflect not only cortisol release, but also altered cortisol clearance from plasma (25,26). In critically ill patients, cortisol levels may vary from less than 5 μg/dL to more than 100 μg/dL and do not reflect the hypothalamic–pituitary–adrenal (HPA) axis function (27). Likewise, ACTH levels may be lower than in noncritically ill patients (26,27), particularly in patients with sepsis (27).

FIGURE 136.3 Integrative approach of the endocrine system. AVP, arginine vasopressin; CRH, corticotropin-releasing hormone; GABA, gamma-aminobutyric acid.

FIGURE 136.4 Signals to the hypothalamus–pituitary axis. There are two different pathways of activation of the hypothalamus–pituitary axis. The humoral pathway acts by (a) cytokines produced in the blood and carried to the hypothalamus and via areas of the brain lacking a blood–brain barrier, (b) cytokines produced by glial cells, and (c) cytokines produced in the pituitary gland and the hypothalamus. The second pathway is neural route. It acts by activation of various afferent neurons of the peripheral system such as vagal fibers. Finally, endogenous gas such as hydrogen sulfide (H2S) or carbon monoxide (CO) may play a role. Refer to Table 136.1 for definitions of abbreviations.
Vasopressin (ADH)
Circulating vasopressin levels are regulated through various stimuli including changes in blood volume or blood pressure, plasma osmolality, cytokines, and other neuromediators (as discussed above). Acute illness may be associated with a syndrome of inappropriate antidiuresis (28). In sepsis, vasopressin levels in plasma may follow a biphasic response with early high concentrations, from abrupt emptying of posterior pituitary stores, followed by a progressive decline over 72 hours to levels indicating relative vasopressin insufficiency, in about one-third of cases (29). The delayed decrease in vasopressin levels in plasma is not related to altered hormone clearance from plasma (29), may be mediated by iNOS overexpression in the parvocellular nuclei (11), or by altered osmoregulation (30).
Thyroid Hormones
Low T3–T4 syndrome has been described for more than 20 years in fasting conditions and in a wide variety of diseases (e.g., sepsis, surgery, myocardial infarction, transplantation, heart, renal and hepatic failure, cancers, malnutrition, inflammatory diseases) and is also called euthyroid sick syndrome or nonthyroidal illness syndrome (NTIS) (31). Early phase of acute stress is associated with decreased serum tri-iodothyronine (T3) levels, increased rT3 levels. Then, serum thyroxine (T4) levels decreased within 24 to 48 hours, and TSH levels remained within normal range without a circadian rhythm (32). Underlying mechanisms included decreased conversion of T4 and T3 in extrathyroid tissues due to inhibition of the hepatic 5′-monodeiodination, lack of substrates due to transport protein inhibitors, loss of thyrotropic negative feedback, cytokines inhibiting thyrotropic centers or expression of thyroid hormone nuclear receptors (33), presence of inhibitory substances such as dopamine. Prolonged critical illness is associated with centrally induced hypothyroidism as suggested by restoration of T3 and T4 pulses by exogenous TRH infusion (34), by diminished thyroid gland weight and follicular size at postmortem examination (35), by reduced expression of TRH mRNA paraventricular nuclei, and low concentrations of tissue T3 (36,37).
Growth Hormone
Acute phase of critical illness is characterized by high levels of growth hormone with low oscillatory activity and low levels of insulin-like growth factor (IGF)-1 (38), suggesting resistance to GH, likely related to decreased expression of GH receptors (39). GH resistance may be beneficial by enhanced direct lipolysis and anti-insulin effects, liberating free fatty acids and glucose to vital organs, and by postponing costly IGF-1 mediated metabolism. In sustained critical illness–related stress, GH secretion showed low pulsatile and high nonpulsatile fractions (40), correlating with low circulating levels of peripheral effectors such as IGF-1. This reduced secretion of IGF-1 did not reflect resistance to GH, but rather, suggested a hypothalamic origin as infusion of GH secretagogues restored a pulsatile GH secretion (41).
Adrenal Medulla Hormones
In resting conditions, very small amounts of adrenaline and noradrenaline are released from the adrenal medulla (i.e., <50 ng/kg/min in the dog). Therefore, removing the adrenal medulla did not affect survival in animals (42). Exposure to stressors like fright, pain, anesthesia, exercise, cold, or endotoxin increased within seconds circulating adrenaline and noradrenaline concentrations by 2 to 3 logs, an effect prevented by removal of the adrenal medulla (43). Then, Cannon introduced the concept of “fight or flight” and established its relationship with catecholamines release. Catecholamine synthesis results from an enzymatic cascade starting from the hydroxylation of the amino acid tyrosine leading to the production of dihydroxyphenylalanine (dopa). Dopa is decarboxylated to form dopamine. Dopamine is then hydroxylated in the side chain to form noradrenaline. Finally, noradrenaline is N-methylated by phenylethanolamine-N-methyltransferase to form adrenaline. Adrenaline is stored in the adrenal medulla in vesicles. Noradrenaline is present in the subcellular granules of the sympathetic nervous endings. Catecholamines have a very short half-life (10 to 20 seconds for adrenaline) and are metabolized through captation, enzymatic inactivation (methylation in liver or kidney; oxidative deamination by monoamine oxidase), or renal excretion. The regulation of catecholamine secretion involves hormonal and nervous factors, and negative feedback through calcium channels. The hormonal regulation depends on cortisol, which is necessary for the enzymatic degradation of catecholamine synthesis. This interaction relies on nerve transmission, paracrine effect, and the local vascular system. The neurogenic regulation involves the cholinergic preganglionic parasympathetic nervous system through the splanchnic nerves. Like cortisol, catecholamine levels in plasma can remain elevated as long as the stress is maintained, even up to a few months after recovery. Nonetheless, obtaining circulating plasma levels of adrenaline and noradrenaline is useless to assess the appropriateness of the catecholamine response to critical illness.
Insulin
Insulin controls glucose metabolism through (i) mobilization of glucose transporters in target cells such as myocytes and adipocytes, (ii) activation of hepatic glucokinase gene transcription, (iii) activation of glycogen synthase and inhibition of glycogen phosphorylase. Other actions of insulin include growth stimulation, cellular differentiation, intracellular traffic, increased lipogenesis, glycogenesis, and protein synthesis. Insulin binds to a ubiquitous membrane receptor of the tyrosine kinase family, IGF-1 and insulin receptor–related receptor (IRR) (44). In acute phase, insulin levels in plasma are rapidly increased as a result of both increased secretion and tissue resistance.
CLINICAL ENDOCRINE-RELATED SYNDROMES DURING CRITICAL ILLNESS
The immediate neuroendocrine activation, mainly sympathoadrenal hormones, results in alertness, insomnia, hyperactivity, pupillary dilation, pilar erection, sweating, salivary secretion, tachycardia, rise in blood pressure with vasodilation of skeletal muscle and coronary arteries, bronchiolar dilation, skin vasoconstriction, mobilization of glucose from liver with hyperglycemia, increased oxygen capacity of the blood via spleen constriction and mobilization of red blood cells, and shortening of coagulation time. However, for the critically ill “escaping” is not possible and fighting stress is the only option, and appropriate neuroendocrine activity to the intensity and duration of stress determines host survival and recovery (Table 136.3). The clinical consequences of the stress-system activation include behavioral changes and cardiovascular, metabolic, and immune adaptations.
Behavioral Syndrome
In animals, infections are associated with anorexia and body weight loss, hypersomnia, psychomotor retardation, fatigue, and impaired cognitive abilities (45). Similar behavioral changes are consistently reported in humans after cytokine or LPS challenge (46). The so-called sickness behavior or depression due to a general medical condition is likely mediated through release of peripheral and brain cytokines. When glucocorticoids and catecholamine responses are insufficient, critically ill patients will develop brain dysfunction that can result to coma.
| TABLE 136.3 Clinical Consequences of Endocrine Activity during Critical Illness | ||
 | ||







Full access? Get Clinical Tree


