168 Endocrine and Metabolic Crises in the Pediatric Intensive Care Unit
Increasing numbers of endocrine and metabolic conditions are being recognized, and the number of children in treatment programs for them is increasing. Although improved screening programs and therapy may decrease the number of children requiring critical care for these conditions, it is likely they will be recognized in increasing numbers of critically ill children for the foreseeable future. There is also increasing awareness of the significance of metabolic changes such has hypo- and hyperglycemia in the pediatric intensive care unit (PICU).1 General principles of PICU management apply to patients with endocrine and metabolic crises (Table 168-1).2,3 Crises may cause damage with long-term sequelae for the child and family; however, they also present unique diagnostic opportunities. The intensivist has a particular responsibility to:
TABLE 168-1 Principles of Management of Metabolic and Endocrine Crises
| Principle | Specifics of Conditions |
|---|---|
| Airway management | Many patients have depressed level of consciousness, and airway management is essential to prevent complications. |
| Breathing support | Acidotic patients may make huge respiratory effort; ventilatory support may help decrease the metabolic demands on these patients. Although administration of sodium bicarbonate may help to settle some of the acidosis-related symptoms such as hyperventilation, bicarbonate may aggravate some problems seen in conjunction with urea cycle defects. Give bicarbonate only if the plasma bicarbonate <10 mmol/L, and then only half correct deficits. |
| Circulatory support | Ensure adequate circulating volume; this may be a particular issue if there has been excessive fluid loss from vomiting or diarrhea. |
| Disability | Control seizures using anticonvulsant agents. Administer pyridoxine if possibility of pyridoxine dependency. |
| Dialysis to remove toxins where necessary | Hemodialysis is the most efficient means of removing toxins such as ammonia and leucine. Hemofiltration is less efficient but may be more applicable in critically ill children. Peritoneal dialysis is slower but has the advantage of ease of initiation.1 In some conditions, it may be possible to remove toxins by stimulating alternative pathways of metabolism. |
| Ensure that glucose is maintained in the normal range | A normal glucose level should be maintained at all times. Excessive administration of glucose in the mitochondrial energy chain problem may exacerbate lactic acidosis. Also, attempt to provide an adequate energy supply (may use medium-chain fatty acids where appropriate). Minimize energy demands on patient. |
| Fluids | In general, provide 1.5× normal fluid maintenance requirements to accelerate excretion of water-soluble toxins. In the context of encephalopathy (MSUD or urea cycle defects), be careful to avoid overhydration, which may contribute to development of cerebral edema. |
| Feeds | If there is accumulation of a product, this needs to be eliminated from the diet (e.g., fructose, galactose). Start with protein-free diet, but do not continue beyond 2 days, because the catabolic state also creates problems. If diagnosis not identified, need gradual reintroduction of feeds and nutrition. If there is deficiency of any nutrient (e.g., carnitine, which may have a primary or a secondary deficiency), supplement that nutrient. Ensure there is an adequate energy source along a metabolic route that is functional. Provide specific vitamin therapy where indicated. |
| Family support and information | The diagnosis of an inborn error of metabolism has major implications for families, and considerable support is required.2 |
| Treat infection | Infections are an important component of pediatric ICU presentation of inborn errors of metabolism. Some conditions such as galactosemia are related to specific infections such as Escherichia coli. Other conditions are related to pyogenic infections because of neutropenia. Children who are in a poor nutritional or metabolic state are more susceptible to infection. Intercurrent infections may be the precipitating factor for metabolic decompensation. |
| Investigations | A wide variety of investigations are relevant to inborn errors of metabolism. Biochemical testing on a range of body fluids and on tissues is fundamental to accurate diagnosis of the problem. Biochemical tests may range from simple screening tests to more complex tests on tissue culture. Imaging techniques such as CT, MRI, magnetic resonance spectroscopy, and echocardiography may be relevant. Functional tests such as EEG, ECG, and EMG may be useful in diagnosis. Increasingly, genetic diagnosis is available if children have recognized genetic mutations. |
| Monitor response to therapy | Clinical monitoring is essential. Biochemical monitoring of the appropriate metabolites is essential to ensure that metabolic control is established. |
CT, computed tomography; ECG, electrocardiogram; EEG, electroencephalogram; EMG, electromyogram; ICU, intensive care unit; MRI, magnetic resonance imaging.
A particular problem of endocrine and metabolic crises is that laboratory investigation of specific conditions may take time while patients require urgent therapeutic intervention. Because it may not always be possible to follow algorithms of investigation, a reasonable approach is to collect all relevant specimens immediately,6 store them appropriately, and liaise with laboratory services to use the specimens in a logical and cost-effective manner to confirm the diagnosis.
 Endocrine Crises
Endocrine Crises
Abnormalities of Glucose Control
Abnormalities of glucose control, including diabetic ketoacidosis, are the most common endocrine crises encountered in the PICU. Hypoglycemia and hyperglycemia are associated with increased mortality1,7 in sick children and may be part of a wide variety of disease processes. Measurement of blood glucose is part of the initial biochemical evaluation of any sick child, particularly if a depressed level of consciousness or shock is present. When an abnormal glucose level has been identified, it must be addressed and levels be remeasured at appropriate intervals until the problem has been resolved. The situation is further complicated by technical issues in the measurement of blood glucose,8,9 with differences between blood and plasma glucose level (glucose concentration in plasma is approximately 11% higher than whole blood because of the higher water content in plasma, but this may be affected by anemia or polycythemia); differences between arterial, venous, and capillary glucose levels (which may also vary depending on clinical context),8 and potentially significant differences between measurement techniques.9 A particular concern is that in general, inaccuracies increase at lower glucose levels.9 Generally, central laboratory devices are taken as the standard, although there is increasing utilization (and convenience) of point-of-care devices.
Hypoglycemia
Hypoglycemia may be associated with devastating damage to the brain and requires immediate attention. In general, a diagnosis of hypoglycemia depends on the presence of symptoms and a low blood glucose level, and resolution of symptoms on correction of the low glucose level. Unfortunately, symptoms of hypoglycemia are relatively nonspecific, ranging from lethargy, poor feeding, hypotonia, and “jitteriness” to convulsions, apneic episodes, cardiovascular collapse, and sudden infant death syndrome (SIDS). Hypoglycemia may be hidden in the complex of critical illness, particularly if patients are deeply sedated and paralyzed. In addition, some diabetic patients have reduced awareness of hypoglycemia.10 Thus, regular monitoring of blood glucose is an important component of the management of any critically ill child.
Hypoglycemia immediately following birth may be common, but there are considerable controversies in the definition of hypoglycemia in this period.11–13 Table 168-2 lists an approach to hypoglycemia immediately following birth.
TABLE 168-2 At-Risk Infants for Whom Routine Monitoring of Blood Glucose Is Recommended
| Associated with Changes in Maternal Metabolism |
| Associated with Neonatal Problems |
| Intrauterine Growth Restriction |
| Hyperinsulinism |
| Endocrine Disorders |
| Inborn Errors of Metabolism |
From Cornblath M, Hawdon JM, Williams AF et al. Controversies regarding definition of neonatal hypoglycemia: suggested operational thresholds. Pediatrics. 2000;105:1141-5.
Symptomatic hypoglycemia occurs more frequently during the neonatal period than in any other period of childhood. Infants at particular risk include infants with poor hepatic glycogen stores (e.g., preterm or small-for-gestational-age infants); poor glucose intake (e.g., preterm or ill infants); and hyperinsulinism, either primary or secondary to high intrauterine glucose levels (e.g., infants of diabetic mothers).14 Hypoglycemia also may be a feature of perinatal illness including asphyxia, polycythemia, hypothermia, septicemia, and respiratory distress syndrome. Much less common causes include growth hormone15 or adrenal insufficiency,16 inborn errors of metabolism, and glucagon insufficiency. Drugs administered to the mother during pregnancy, including oral hypoglycemic agents, also must be considered.
In childhood, hypoglycemia may result from inadequate glucose intake (prolonged starvation, malabsorption); defects in glycogenolysis (glycogen storage disorders) or gluconeogenesis (fructose-1,6-diphosphatase deficiency, ethanol intoxication, Jamaican vomiting sickness, etc.), fatty acid oxidation disorders and defects in ketogenesis, deficiency of gluconeogenic hormones (e.g., adrenalin, corticosteroids, glucagons, growth hormone, thyroid hormone), excessive insulin secretion (hyperinsulinism), and a variety of specific disorders including abnormalities of amino acid metabolism.17
As soon as hypoglycemia is noted, specimens should be collected immediately for appropriate tests (Table 168-3). Treatment of hypoglycemia with intravenous (IV) glucose should be initiated promptly. An initial bolus dose of 0.5 g/kg of glucose (may need 0.5-2 g/kg in neonates) should be given as a 10% or 25% (in older children) dextrose solution, followed by an ongoing infusion of glucose at a rate of 4 to 8 mg/kg/min. The concentration of the ongoing infusion depends on the fluid requirements of the child and the availability of central venous access (for higher concentrations). Glucagon may be given at a dose of 0.1 to 0.3 mg/kg (IV or intramuscularly [IM]) but is unlikely to be effective in patients with low glycogen stores, glycogen storage disorders, or hepatic dysfunction. Hydrocortisone at a dose of 5 mg/kg every 12 hours may be useful in some patients. Diazoxide and IV octreotide decrease insulin release and may be useful in the management of hyperinsulinemia.
TABLE 168-3 Investigation of Hypoglycemia
| Blood glucose | Measurement of glucose using blood from capillary specimens and using test strips may be unreliable (particularly in poorly perfused patients or patients with high hematocrit); where possible, low glucose levels should be confirmed using laboratory assays on venous or arterial blood. |
| Actual glucose intake | Hypoglycemia in the presence of normal glucose intake or after brief fast suggests hyperinsulinism. Hypoglycemia after hours of fasting is associated with fatty acid oxidation defects and endocrine insufficiency. |
| Non–glucose-reducing substances in the urine | Particularly in neonates and probably not relevant in older children. If present in the urine, consider galactosemia, hereditary fructose intolerance, or tyrosinemia. |
| Serum and urinary ketones | Low ketones suggest hyperinsulinism or fatty acid oxidation problem. |
| Serum free fatty acids | Free fatty acids are low in hyperinsulinism but high in fatty acid oxidation defect. |
| Serum insulin (and C peptide), cortisol, glucagon, growth hormone, and thyroid levels | Normal serum insulin in the presence of hypoglycemia is evidence of hyperinsulinism. C peptide may be necessary to ascertain whether exogenous insulin was administered. Release of C peptide may not be as pulsatile as that of insulin. |
| Serum ammonia | To recognize hyperinsulinism/hyperammonemia syndrome |
| Urinary organic acids and serum amino acids | To diagnose fatty acid oxidation defects (urinary organic acids). Aminoacidopathies such as MSUD, propionic acidemia, isovaleric acidemia, methylmalonic acidemia, and tyrosinemia may also present with hypoglycemia. |
| Total and free carnitine with acylcarnitine profile | To recognize primary and secondary deficiency of carnitine and fatty acid oxidation defects |
Neonates
In the neonatal period, glucose is not the only energy source from oxidative metabolism in the brain, and alternative energy sources such as ketones may be used.11 In fact, breast-fed babies routinely have lower glucose levels and higher ketone levels than formula-fed infants. Recent reviews have highlighted that “there is inadequate information in the literature to define any one value of glucose below which irreparable hypoglycemic injury to the central nervous system occurs, at any one time or for any defined period of time, in a population of infants or in any given infant.”18 However, there is evidence that hypoglycemic injury is more likely to occur at very low levels of glucose (20-25 mg/dL [1.1-1.4 mmol/L]) and if hypoglycemia is prolonged, is the consequence of hyperinsulinemia (when alternative energy sources for the brain may be very limited), and in the presence of other potential injuries.18,19
A suggested approach to hypoglycemia in the neonatal period is shown in Figure 168-1. Although the threshold for treatment in the asymptomatic neonate is 25 to 30 mg/dL (1.1-1.4 mmol/L), the recommended levels during treatment are above 45 mg/dL (2.5 mmol/L).
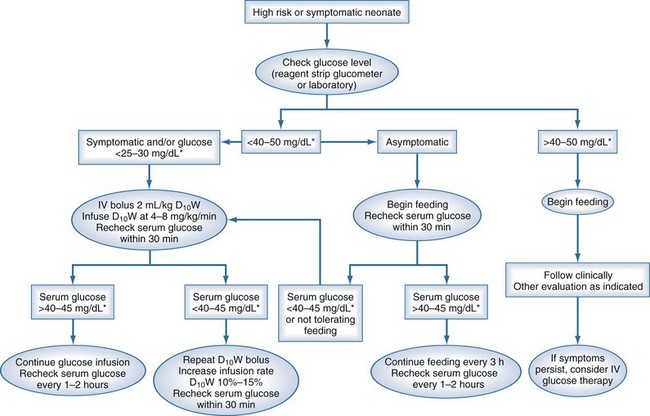
Figure 168-1 A suggested approach to management of the neonate with low glucose.
(From Rozance PJ, Hay WW. Hypoglycemia in newborn infants: features associated with adverse outcomes. Biol Neonate 2006;90:74-86.)
Although the exact definition of hypoglycemia in children is controversial, a minimal level of 2.6 mmol/L or greater should be maintained to ensure normal neural function.12,20,21 It probably is safer to maintain a level of greater than 3.5 mmol/L. Because there are multiple causes for hypoglycemia, and symptoms may not be due to the hypoglycemia alone, it is essential to identify the cause.
Hypoglycemia is associated with severe illness. A wide range of illnesses including infections,22 cyanotic and acyanotic congenital heart disease, and cardiomyopathy/myocarditis have been associated with hypoglycemia. Hepatic failure from infection, toxin ingestion, or drug reactions may be associated with severe hypoglycemia, and Reye syndrome classically presents with hypoglycemia. Toxins such as salicylates and ethanol also may cause hypoglycemia. Hypoglycemia has been linked with increased mortality from malaria,23–26 gastroenteritis,27 and acute bacterial meningitis28 among other conditions. Hypoglycemia also has been described as a complication of therapy for leukemia with mercaptopurine and methotrexate.29,30 Although severe illness or sepsis may be an adequate explanation for hypoglycemia, a diagnosis of sepsis should not exclude the possibility of an endocrine or metabolic crisis.
Hyperinsulinemic Hypoglycemia
Hyperinsulinism is the most common cause of persistent or recurrent hypoglycemia in infancy.31–33 Hyperinsulinism may be secondary to risk factors in the perinatal period (associated with high maternal glucose levels,34 rhesus incompatibility, intrauterine growth retardation,35 and perinatal asphyxia36) but may also be congenital37,38 or associated with Beckwith-Weidemann syndrome and some other developmental syndromes.33
Although most patients with hyperinsulinemic hypoglycemia present in the neonatal period, first presentation may be during infancy and occasionally during childhood,39 when the condition may be more likely to respond to medical therapy. Neonates with hypoglycemia may have the macrosomia typical of infants of diabetic mothers, but hyperinsulinemic hypoglycemia may occur in apparently normal infants of normal or low birth weight. Hypertrophic cardiomyopathy and hepatomegaly may be seen31 in affected infants. The characteristic features of hyperinsulinism include hypoglycemia with glucose requirements of greater than 6 to 8 mg/kg/min to maintain normoglycemia, absence of ketonemia and ketonuria, low plasma free fatty acids and branch chain amino acids, detectable insulin at the time of hypoglycemia, and response to glucagon administration.31 The combination of hypoglycemia with low free fatty acids and absence of ketonemia is responsible for the potentially devastating effects of this condition on the brain, as it is deprived of both normal and alternate substrates.32
Hyperinsulinemic hypoglycemia with hyperammonemia (previously called leucine-sensitive hypoglycemia) is well described40,41 and is attributed to mutations in the gene for glutamate dehydrogenase. Patients generally respond well to therapy with diazoxide, and consumption of extra carbohydrate before protein meals may help ameliorate symptoms. Special low-leucine milks are available.
Congenital hyperinsulinemic hypoglycemia is caused by abnormalities in genes controlling the secretion of insulin by the beta cells of the pancreas, with abnormalities described in seven genes.32
Clinically, hyperinsulinemic hypoglycemic patients may be categorized by their response to diazoxide (5-20 mg/kg/d in 2-3 divided doses), with most responding. Exceptions include those with congenital hyperinsulinemia related to focal hyperinsulinemia and those with diffuse hyperinsulinemia related to inactivating mutations in ABCC8 and KCNJ11. Unfortunately, diazoxide may predispose to fluid retention, and use must be carefully monitored. Chlorothiazide (7-10 mg/kg/day in 2 divided doses) may be added (particularly in neonates).32 Nifedipine (0.25-2.5 mg/kg/d in 3 divided doses) may also be useful in some patients.42
A suggested approach to ongoing diagnosis and management is outlined in Figure 168-2, showing a marked change from previous practice. In those patients who are responsive to diazoxide, that will remain the basis of therapy. In those with no response to diazoxide, genetic testing (for homozygous or compound heterozygous mutations in ABCC8 and KCNJ11), followed by fluorine-18 (18F)-dopa positron emission tomography (PET) scanning for those with potentially focal pancreatic lesions will enable identification of those who may benefit from resection of the pancreas. Pancreatic islets cells take up L-3, 4-dihydroxyphenylalanine (L-dopa), where it is converted to dopamine by dopa-decarboxylase. Uptake of the positron-emitting tracer 18F-dopa PET is increased in beta cells with a high rate of insulin synthesis and secretion provides visualization of the focal lesion.43–46 Patients with focal lesions should respond to partial pancreatectomy, which may be done laparoscopically.47,48 Diffuse disease that is unresponsive to diazoxide therapy will require a near-total pancreatectomy and may be associated with a high incidence of both endocrine and exocrine problems.49
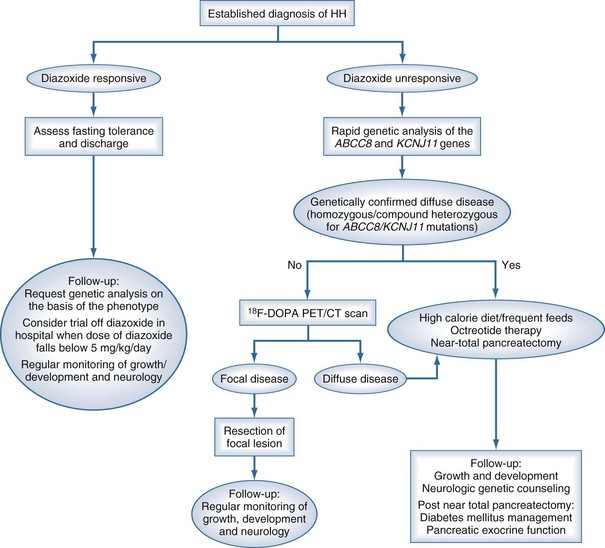
Figure 168-2 Flow chart outlining the management cascade of neonates with hyperinsulinemic hypoglycemia (HH). Clinically, HH can be classified into diazoxide-responsive and diazoxide-unresponsive disease. A fluorine-18 L-3, 4-dihydroxyphenylalanine positron emission tomography (18F-dopa PET) scan is currently only indicated in neonates who are unresponsive to diazoxide and do not have genetically confirmed diffuse disease.
(From Kapoor RR, Flanagan SE, James C, Shield J, Ellard S, Hussain K. Hyperinsulinaemic hypoglycaemia. Arch Dis Child 2009;94:450–7.)
Close long-term follow up will be required in all these patients, and there may be significant neurologic and psychological problems to be dealt with.49
Ketotic Hypoglycemia
Although ketotic hypoglycemia (“accelerated starvation”) is probably the most common cause of hypoglycemia in previously healthy children,50 it is unlikely to present in the PICU. This condition usually affects children aged 6 months to 8 years, and the clinical features include ketosis, severe nausea, and hypoglycemia, usually occurring in the morning after a moderate fast. Treatment consists of ensuring that there is an adequate and regular intake of glucose, particularly during intercurrent infections. Urinary ketones may act as a warning signal, because the ketosis usually precedes the onset of hypoglycemia by several hours.
Adrenal Insufficiency
Adrenal insufficiency after high-dose inhaled corticosteroid therapy has presented with hypoglycemia51–54 and should be considered if there is a past history of inhaled steroid use (particularly fluticasone).53,55 Adrenal insufficiency also may occur after adrenal bleeds (e.g., after meningococcal septicemia or difficult delivery), as part of adrenal disease (e.g., congenital adrenal hyperplasia or hypoplasia) in which ambiguous genitalia may (or may not) be a pointer in females, or as part of hypopituitarism (e.g., congenital, after craniopharyngioma, or after cranial irradiation56). Some patients with primary adrenal insufficiency may present with hypoglycemia, particularly during acute illnesses.16,57 Adrenoleukodystrophy should be considered as part of the etiologic diagnosis in any male patient with Addison’s disease (pigmentation may be a clue) and should be tested for by measurement of very-long-chain fatty acids.58,57
There has been considerable interest in adrenocorticoid deficiency in children with critical illness and particularly acute severe sepsis (see Chapter 131).59–63 Currently, supplementary steroids are recommended for children with acute severe sepsis and catecholamine resistant shock,64,65 but there are not clear definitions for either adrenal insufficiency or catecholamine resistance, nor are there firm recommendations for the dose of adrenal replacement therapy.
Adrenocorticoid deficiency also has been shown in preterm infants.66 A randomized controlled study of “stress” dose hydrocortisone therapy in hypotensive very low-birthweight infants showed that steroids were effective in treating refractory hypotension.67
Congenital adrenal hyperplasia is associated rarely with hypoglycemia. Female patients are usually diagnosed early in life as a result of virilization, whereas male patients tend to present later. Patients with the salt-losing form of congenital adrenal hyperplasia present with hyponatremic dehydration and shock, usually associated with hyperkalemia. Because patients with salt-wasting 21-hydroxylase deficiency also may have catecholamine deficiency, shock may be a significant feature. Diagnosis is based on the clinical picture, typical electrolyte pattern, hypoaldosteronism, and hyperreninemia.68 Long-term treatment consists of hydrocortisone (to suppress excess secretion of corticotropin-releasing hormone and corticotropin), 10 to 20 mg/m2 of body surface area per day in three divided doses, although larger doses may be required during adrenal crises, together with mineralocorticoid replacement (0.1-0.2 mg of fludrocortisone daily) and sodium chloride supplementation. Little is known about the dose of hydrocortisone required during critical illness, although Charmandari et al.69 showed that when 6-hourly bolus doses of 15 mg/m2 of hydrocortisone are given, high immediate serum levels are achieved, followed by rapid decline to undetectable levels by 4 hours after administration. These authors postulated that continuous infusion of hydrocortisone may be more appropriate in critical illness.
Growth Hormone Deficiency
In the neonatal period, growth hormone deficiency presents with hypoglycemia (possibly with seizures), prolonged jaundice, and in boys, micropenis and undescended testes. Growth failure becomes apparent only toward the end of the first year of life. In later childhood, growth failure is a more common presentation, and hypoglycemia rarely occurs56 unless associated with adrenocorticotropic hormone deficiency.
Hyperglycemia Other Than Diabetes Mellitus
Hyperglycemia is relatively common in the PICU.70 In a retrospective study of 948 nondiabetic patients admitted to the PICU, there was a high prevalence of hyperglycemia, with 70.4% of patients having a glucose value above 120 mg/dL, 44.5% above 150 mg/dL, and 22.3% above 200 mg/dL within 10 days of admission. A 2.5-fold increased risk of dying was seen if the maximum glucose obtained within 24 hours of admission was over 150 mg/dL and a 5.68-fold increased risk if the maximum glucose obtained within 10 days of admission to the PICU was over 120 mg/dL.71 However, that study was retrospective and not corrected for severity of illness. In addition, ascertainment bias was present,72 so the study could not really provide insight in terms of causality or the potential impact of therapy.73
Hyperglycemia is common in a wide variety of conditions including bronchiolitis,74 sepsis, hemolytic uremic syndrome,75 tetanus,76 and toxin ingestion (e.g., theophylline poisoning77). Other studies have confirmed that high glucose levels are not only common in the PICU population but are associated with increased mortality and/or morbidity in a wide variety of conditions.78–81
An initial report from an adult surgical unit (predominantly cardiac)82 provided evidence that “tight” control of glucose levels was associated with a significant improvement in patient outcomes. The same group studied a cohort of patients in a medical ICU, and there was no difference in mortality between groups.83 There have been numerous studies in a variety of adult critical care populations since that time, with positive effects of tight glucose control noted on cholestasis,84 renal function,85 neurologic and neuromuscular complications,86,87 and endothelial function.88 Unfortunately, there have also been increased reports of iatrogenic hypoglycemia, and a recent meta-analysis of studies in adults89 concluded that tight glucose control was not associated with an improvement in hospital mortality and was associated with an increased incidence of hypoglycemia. Subsequently, a large randomized controlled trial (RCT) of adult patients compared “tight” (81-108 mg/dL or 4.5-6.0 mmol/L) with “conventional” glucose control (target of ≤180 mg/dL or ≤10.0 mmol/L). The 90-day mortality was higher in the group on tight glucose control, and subgroup analysis showed that the outcomes favored conventional control in all groups except trauma patients and patients on steroids.
Pediatricians have been more cautious in their approach to control of hyperglycemia, but a number of protocols for PICU management of hyperglycemia have been implemented and reported.90,91 An RCT of protocols for tight glucose control in the PICU showed no difference between a paper-based protocol and a computerized decision support tool.92
In a study of glycemic control in 177 postoperative cardiac patients,93 there was no difference in glucose levels on day 1 between survivors and nonsurvivors, but the 5-day mean peak glucose levels were significantly higher in nonsurvivors. Insulin usage was higher in the nonsurvivors, and nonsurvivors had more hypoglycemic events. The authors speculated that targeting a more permissive glucose level of 90-140 mg/dL (5-7.7 mmol/L) might be associated with both improved outcomes and reduced risk of hypoglycemia. In a retrospective review of 100 postoperative cardiac patients, there was high incidence of hyperglycemia (and an association with higher severity of illness), and implementation of a pediatric glycemic control protocol had a low incidence of hypoglycemia.79
A prospective RCT of 700 critically ill children (317 infants and 383 children) admitted to PICU94 randomized patients to targeted blood glucose levels (throughout PICU stay) of 2.8 to 4.4 mmol/L in infants and 3.9 to 5.6 mmol/L in children, with insulin infusion throughout PICU stay (intensive group [n=349]) or to insulin infusion only to prevent blood glucose from exceeding 11.9 mmol/L (conventional group [n=351]). Mean blood glucose concentrations were lower in the intensive group than in the conventional group, and hypoglycemia (glucose ≤2.2 mmol/L) occurred in 87 (25%) patients in the intensive group (P < 0.0001) versus 5 (1%) patients in the conventional group. Severe hypoglycemia (blood glucose less than 117 mmol/L) occurred in 17 (5%) of the intensive group versus 3 (1%) of the conventional group (P=0.001). Duration of PICU stay was reduced in the intensively treated group (5.51 days [95% CI, 4.65-6.37] versus 6.15 days [95% CI, 5.25-7.05]; P=0.017). The number of patients with stay in PICU longer than the median was 132 (38%) in the intensive group versus 165 (47%) in the conventional group (P=0.013). Nine (3%) patients died in the intensively treated group versus 20 (6%) in the conventional group (P = 0.038). There is ongoing debate about appropriate targets for glucose, optimization of protocols, balance of nutrient and glucose intake versus insulin therapy, and the like.
In the context of major burns there is also some evidence that insulin therapy to maintain lower blood glucose levels may be associated with improvements in metabolism.95
A recent review96 concluded:
Pediatricians have been reluctant to implement tight glucose control in PICU because of concerns about the deleterious effects of hypoglycemia.97,98
A recent review of hyperglycemia in the preterm infant99 suggested the following pragmatic approach to management: confirm hyperglycemia with laboratory test; treat any underlying problem such as sepsis, stress, etc.; calculate glucose infusion rates, and if above 12 mg/kg/min, reduce infusion rate; treat with insulin if glucose is over 10 mmol/L (or other symptoms such as polyuria), but start cautiously with very low doses; finally, if hyperglycemia persists, consider other diagnoses such as diabetes.
Diabetes Mellitus
Children with diabetes mellitus have a higher mortality than healthy children,100,101 with standardized mortality ratios of 2.15102 to 4.2,103 although some deaths are not directly related to diabetes. The highest mortality is in children aged 1 to 4 years, in whom the standardized mortality ratios may be 9.2104 to 13.7.105 Most deaths attributable to diabetes mellitus occur as a consequence of diabetic ketoacidosis (DKA) or hyperglycemia, with the remainder attributable to hypoglycemia.104 Diabetic ketoacidosis is relatively common at the time of first presentation, particularly in younger children,106 in whom diagnosis may be delayed. Although the incidence of type 1 diabetes mellitus has been increasing in many parts of the world, the hospitalization rate for DKA in established and new cases of type 1 diabetes mellitus has not increased in Canada and Europe since the 1990s107,108 because of earlier diagnosis and safer ambulatory management with the help of a multidisciplinary team.
The mortality rate in the developed world for DKA ranges from 0.15% to 0.31%109 but may be far higher in other settings.110 The most common cause of death among patients with DKA is cerebral edema. Other causes of death in DKA include electrolyte disturbances, hypoglycemia, pulmonary edema, rhabdomyolysis, infections (including mucormycosis), and thrombosis. The management of DKA in childhood has been extensively reviewed elsewhere,109,111,112 with current recommendations.
Cerebral Edema in Diabetic Ketoacidosis
In affluent countries, symptomatic cerebral edema occurs in 0.5% to 1% of pediatric DKA episodes,113 with risks being higher in young children and previously undiagnosed diabetics. Mortality is high (21%-24%), and 15% to 26% of survivors will have permanent morbidity (including pituitary insufficiency).113
The exact mechanisms of cerebral edema in DKA are not clear,114 although some imaging studies suggest that cerebral edema may be related to vasogenic factors rather than osmotic factors.115 Cerebral hyperemia has also been demonstrated as part of abnormal autoregulation.116,117 Factors that have been associated with the development of cerebral edema include the administration of bicarbonate, a higher plasma urea, arterial partial pressure of carbon dioxide (PCO2),118,119 and a smaller increase in plasma sodium concentration during therapy.120 However, a recent systematic review121 of the literature concluded that there was no clear evidence that treatment was related to the development of cerebral edema. Cerebral ischemia and reperfusion injury have also been considered.122 Cerebral edema may be present before therapy for DKA in 5% of cases, although most cases develop 4 to 12 hours after initiation of therapy.118,123 The clinical signs of cerebral edema in DKA are variable and include headache, deterioration in level of consciousness, inappropriate slowing of pulse rate, and increased blood pressure. However, children with no clinical signs of cerebral edema have been documented to have brain swelling,124 and a significant proportion of children have disrupted memory function following episodes of DKA.125
Adverse outcomes have been associated with greater neurologic depression at the time of diagnosis, high initial serum urea nitrogen,118,126 and intubation with hyperventilation to a PCO2 less than 22 mm Hg.126,127
Although the biochemical derangements of hyperglycemia, metabolic acidosis with ketosis, and electrolyte abnormalities are the most obvious problems in DKA, significant derangements in other systems have been documented, including plasma tryptophan levels,128 thiamine levels,129 cytokine130 and lymphocyte responses,130 and coagulation abnormalities.130 There is little doubt that DKA is associated with a thrombotic state131 and an increased incidence of cerebrovascular accidents, and care should be taken about the use of femoral central venous access because this may have a higher than usual complication rate in these patients.132 A reported case of myocardial infarction related to DKA133 may be a complication of the thrombotic state. Although myocardial function is generally normal in DKA, myocarditis134 has been noted in occasional case reports, whereas pulmonary edema may be more common than previously recognized.135 Prolongation of the QTc interval may be common in DKA (it correlates with ketosis), and careful cardiac monitoring is essential.136
Principles of Management
Management of DKA should be coordinated by an experienced diabetes team. The biochemical criteria for the diagnosis of DKA include a serum glucose concentration above 11 mmol/L (~200 mg/dL), ketonemia and ketonuria, and acidosis with venous pH below 7.3, or serum bicarbonate level below 15 mEq/L.112 The severity of DKA is defined by the level of acidosis, with mild having venous pH less than 7.3 (or bicarbonate <15 mmol/L); moderate, pH less than 7.2 (or bicarbonate <10 mmol/L); and severe, pH less than 7.1 (or bicarbonate <5 mmol/L).112 Children with severe DKA should be managed in a specialized diabetic unit or in the PICU.
Baseline Assessment
An admission weight should be obtained if at all possible, and future therapy should be based on this weight. Blood samples should be taken for the following investigations: serum or plasma glucose, electrolytes (including bicarbonate or total carbon dioxide), blood urea nitrogen, creatinine, osmolality, venous (or arterial in critically ill patient) pH, PCO2, calcium, phosphorus, and magnesium concentrations (if possible), HbA1c, hemoglobin and hematocrit or complete blood count. Measurement of blood β-hydroxybutyrate concentration, if available, is useful to confirm ketoacidosis and may be used to monitor the response to treatment.137–140 Urine specimens should be analyzed for ketones. Electrocardiograms may be useful if delays are expected in getting potassium results.
Fluid Management
The objectives of fluid and electrolyte replacement therapy are restoration of circulating volume, replacement of sodium and body fluid deficit, improved renal function with enhanced clearance of glucose and ketones from the blood, and minimization of risk of cerebral edema.112 There is a wide range in the amount and rate of fluid and electrolyte loss in patients presenting with DKA (depending on the rate of onset and duration of symptoms, the severity of vomiting or diarrhea or both, and the fluid ingested by the patient).112 There is a wide range of intravascular status ranging from normovolemia to severe hypovolemia (uncommon). Clinical assessment of dehydration is notoriously inaccurate, and there is an unpredictable rate of ongoing fluid loss related to the osmotic diuresis. In the (unusual) presence of hypovolemic shock, it is reasonable to infuse 0.9% saline using aliquots of 5 to 10 mL/kg until an acceptable blood pressure is obtained.141 Typically, 10 to 20 mL/kg needs to be infused over 1 to 2 hours.109 Ringer’s lactate may be a reasonable alternative, because administration of large volumes of 0.9% saline has been associated with the development of hyperchloremic acidosis. There is no evidence to support the use of colloid solutions.
Thereafter the acceptable principles are that hypovolemia, rapid changes in plasma osmolality, and large volumes of sodium uptake should be avoided. Fluid therapy should be calculated to achieve rehydration over 48 hours.109,112,113 Careful monitoring of fluid balance is essential to ensure that patients are neither losing excessive fluid (via osmotic diuresis) nor gaining excessive fluid. Fluid with a tonicity less than that of 0.45% saline should not be used, and a positive balance of around 6 mmol of sodium chloride per kilogram over 24 hours should be regarded as the upper limit.141 The rate of fluid infusion rarely exceeds 1.5 to 2 times the usual daily requirement.
Despite the fact that almost all patients with DKA are potassium depleted, serum potassium levels frequently are increased at presentation. With initiation of insulin therapy and correction of acidosis, there is rapid intracellular movement of potassium, and careful monitoring of potassium levels is essential. As soon as potassium levels are less than 5.5 mEq/L, 30 to 40 mEq/L of potassium should be added to the fluid infusions, and 0.5 to 1 mEq/kg/h of potassium may be required to correct potassium deficits. Potassium may be given as chloride or phosphate. Although severe hypophosphatemia is relatively common,142 and symptomatic hypophosphatemia has been reported,143 there is no evidence that phosphate administration is routinely necessary in the management of DKA, and the clinical effects of severe hypophosphatemia rarely are seen in DKA. Theoretically, phosphate administration may reduce insulin resistance and depletion of adenosine triphosphate and have positive effects on 2,3-diphosphoglycerate.144 Administration of potassium phosphate helps decrease the chloride load given to patients with DKA. Potassium phosphate may be used safely,145 provided that calcium levels are monitored carefully.146,147 Glucose must be added to the infusion of fluids when the glucose levels are 14 to 17 mmol/L to avoid hypoglycemia.
Bicarbonate
The use of bicarbonate in DKA is extremely limited. Many studies have shown no clinical benefit from its administration.148–150 More recently, bicarbonate administration has been associated with the development of cerebral edema. It should not be given routinely, not in bolus form, and possibly only in patients who have a pH of less than 6.9 despite appropriate correction of intravascular volume and ongoing adequate insulin therapy.



Full access? Get Clinical Tree


