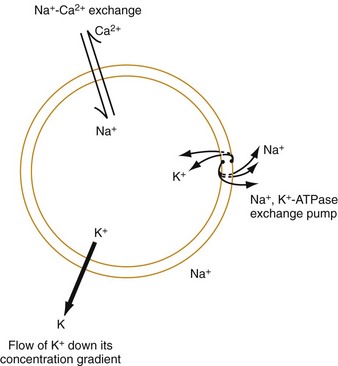
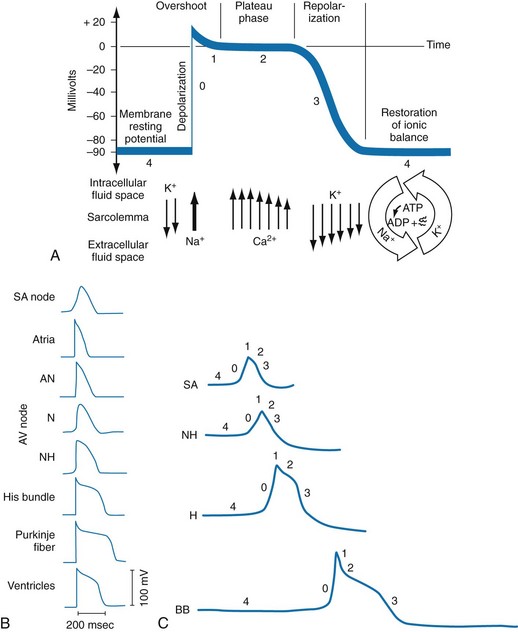
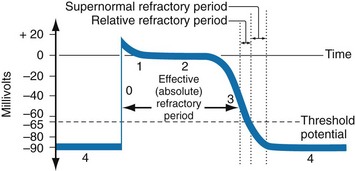
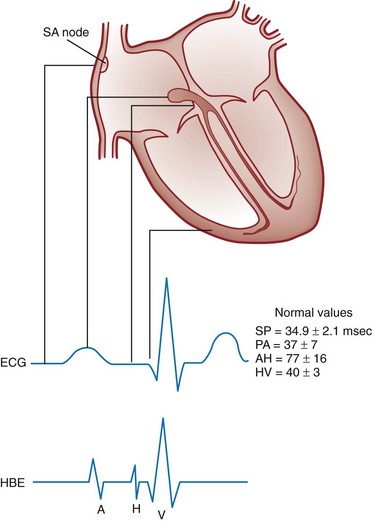
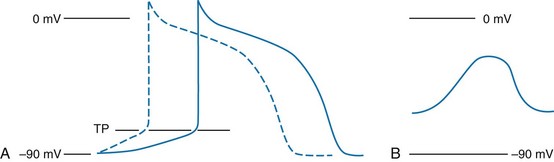
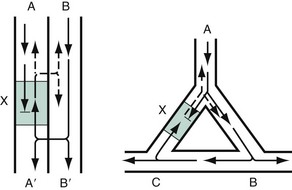
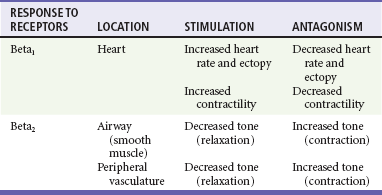
Chapter 79 The function of individual cells in the conductive and contractile tissues of the heart depends on an intact resting membrane potential. Na+, K+, and Ca2+ ions create the membrane potential and regulate conduction and contractility. The membrane potential is the result of a differential concentration of Na+ and K+ on either side of the cell membrane, measuring approximately −90 mV in normal resting, nonpacemaker cells. This electrical gradient exists mainly because of the Na+-K+ exchange pump and the natural concentration-dependent flow of K+ out of the cell. Adenosine triphosphate (ATP) fuels Na+ transport out to the extracellular fluid, with Mg2+ used as a cofactor (Fig. 79-1). This process creates an osmotic gradient, allowing Ca2+ to be passively exchanged for Na2+ and promoting conduction as well as myofibril contraction. The resting membrane potential is generated from the flow of K+ down a concentration gradient toward the extracellular fluid. The cell membrane is far more permeable to potassium than sodium ions, resulting in a net loss of intracellular positive charge. In normal nonpacemaker cells, the application of an electrical stimulus causes the membrane potential to become less negative, termed depolarization. When the membrane potential reaches −70 mV, specialized Na2+ channels open, causing a rapid influx of positive charge into the cell. This “fast” channel activity further decreases the membrane potential and is augmented at 30 to 40 mV by a second “slow” channel that allows Ca2+ influx. When these channels close, resting potential is restored by the sodium-potassium pump, an event known as repolarization (Fig. 79-2). In nonpacemaker cells, additional depolarization from a second electrical stimulus is not possible when the membrane potential is more positive than −60 mV. This period is termed the effective refractory period (Fig. 79-3). At a membrane potential of −60 to −70 mV, a strong impulse can cause a response that is likely to be propagated, although abnormally; this response represents the relative refractory period. At a membrane potential of −70 mV or less, virtually all fast channels are ready for activity if properly stimulated (see Fig. 79-3). Figure 79-4 correlates the surface ECG tracings with tissue electrical events. Impulses generated from within the SA node itself, imperceptible on the surface ECG, are propagated through the atrial tissue to the AV node. Atrial depolarization is characterized by the P wave on the surface ECG. The AV node is an area of specialized tissue between the atria and the ventricles of the heart, located in the posterior-inferior region of the interatrial septum. Its blood supply is from a branch of the RCA in 90% of patients (right dominant) and from the LCA in the remaining 10% (left dominant). Transmission of impulses within the AV node is slower than in parts of the conducting system (Table 79-1) because of a dependence on slow-channel ion influx for membrane depolarization. In some patients, pathologic “accessory pathways” connect atrial and ventricular tissues. These accessory pathways do not share the normal conduction delay of the AV node and may allow for rapid ventricular rates. Preexcitation refers to the early depolarization of ventricular myocardium when accessory paths are used instead of the normal conduction system. There are three electrophysiologic mechanisms: enhanced automaticity, triggered activity, and reentry. Enhanced automaticity includes spontaneous depolarization of nonpacemaker cells or a lower threshold for depolarization in normal pacemaker cells (Fig. 79-5). Both types of enhanced automaticity can occur in the setting of ischemia, electrolyte disturbances, or drugs. Examples of enhanced automaticity include idioventricular rhythms in the setting of acute myocardial infarction, and atrial tachycardias or junctional tachycardias (JTs) seen with digitalis toxicity. Reentry dysrhythmias occur as a consequence of abnormal conduction (Fig. 79-6). For a reentry mechanism, two alternate pathways for conduction must be present and one path must have a longer refractory period. The unequal responsiveness of the limbs creates a functional unidirectional block such that when the impulse exits one limb, it may then reenter the other in retrograde fashion. The cycle is then repeated, creating a self-sustaining or “circus movement” tachycardia that can appear orderly or disorderly (i.e., fibrillatory). Medications used to treat dysrhythmias are classified into four major categories on the basis of their electrophysiologic effects (Box 79-1). Other agents fall outside this classification system and are discussed separately. All antidysrhythmics can cause “prodysrhythmic effects.” This occurs most often in patients with existing structural heart disease and in patients receiving new or higher doses of antidysrhythmic agents. The class I and III agents are associated with prodysrhythmic effects in up to 15% of patients.1,2 The class IC agents profoundly slow depolarization and conduction and have pronounced antidysrhythmic properties.1,2 Up to 15% of patients treated with class IC agents experience new or increased ventricular dysrhythmias. Class IC agents are approved only for oral use in the United States. Side effects of beta-blockers include bronchospasm, bradycardia, hypotension, and heart failure. All beta-blockers are active at both beta1 and beta2 receptors (Table 79-2) to varying degrees. Those with more prominent beta1 effects are said to be cardioselective. Relative contraindications to the use of beta-blockers include asthma or chronic obstructive lung disease, advanced congestive heart failure, and third-trimester pregnancy. Beta-blockers should not be used in patients with preexisting bradycardia or heart block beyond first-degree. Intravenous beta-blockers should be used cautiously in conjunction with calcium channel blockers because of the risk of additive side effects. Acute side effects of beta-blockers include bronchospasm, heart failure, excessive bradycardia, and hypotension. Table 79-2 Cardiac and Respiratory Beta-Adrenergic Receptors and Responses to Pharmacologic Manipulation All class III agents prolong the refractory period by blocking K+ channels, and they have variable effects on the QT interval.1–4 In general, class III agents are alternatives to the class I agents for the treatment of many ventricular and atrial dysrhythmias. The serum half-life of amiodarone is 25 hours after a single intravenous dose and up to 50 days during long-term oral use. Because of the unusual pharmacokinetics, oral regimens vary widely. The acute side effects of amiodarone are primarily limited to hypotension, bradycardia, and heart failure (Box 79-2). There is an additive risk of bradycardia and hypotension when amiodarone is used in conjunction with calcium channel or beta-adrenergic blockers. Rates of prodysrhythmia are relatively low (1-3%). Long-term amiodarone use, however, is associated with significant extracardiac side effects, including irreversible lung and thyroid disease. Amiodarone alters the pharmacokinetics of numerous other drugs, including digoxin and warfarin. Ibutilide has a unique mechanism of action characterized by induction of a slow inward Na2+ current, thereby prolonging the refractory period. Intravenous ibutilide is approved for cardioversion of atrial fibrillation and atrial flutter.5 Ibutilide is associated with QT interval prolongation, and polymorphic VT is more common than with amiodarone although still relatively rare (0.9-2.5%). Dofetilide is a pure class III agent that blocks K+ efflux, increasing the refractory period. It is approved for chemical cardioversion and maintenance of sinus rhythm in patients with atrial fibrillation or flutter,6 but because of its prodysrhythmic effects, it has limited usefulness. Structurally related to amiodarone, dronedarone displays class III properties plus those of other antidysrhythmic classes. The incidence of polymorphic VT associated with dronedarone is very low. Dronedarone is approved for oral use to maintain sinus rhythm in patients with atrial fibrillation or flutter but is contraindicated in patients with severe or recent heart failure decompensation.7 Long-term effects and the ideal populations for the use of this drug remain unclear.8 Digoxin (0.25-0.5 mg IV) can control the ventricular rate in patients with SVTs, including atrial fibrillation and atrial flutter. Because of its delayed onset of action and narrow therapeutic window, however, digitalis is not a first-line agent for emergency therapy. It is not true that digoxin promotes conversion to a sinus rhythm any more than other rate-controlling agents.9 Side effects of digoxin are listed in Box 79-3 and are aggravated by hypokalemia, hypercalcemia, hypomagnesemia, increased catecholamines, and acid-base disturbances. Digoxin overdose therapy is covered elsewhere. Adenosine is a naturally occurring purine nucleoside used for the termination of narrow-complex tachydysrhythmias. Administered as an intravenous bolus, adenosine causes an abrupt slowing of AV conduction in both anterograde and retrograde pathways. Adenosine usually has an onset of action of 5 to 20 seconds and a duration of effect of 30 to 40 seconds. Except in rare cases of catecholamine-induced ventricular dysrhythmias, adenosine has little or no effect on infranodal conduction. For this reason, adenosine is often used as a diagnostic agent in patients with wide-complex tachydysrhythmias when the cause is unclear.10–12 An initial dose of 6 mg as a rapid bolus for adults weighing 50 kg or greater is recommended, with flush through a large peripheral vein. If no response is seen within 1 to 2 minutes, the second dose is doubled (12 mg) and administered. If no effect is seen after a final 12-mg dose, the rhythm should be reassessed and another agent used. Pediatric doses are 0.05 mg/kg initially with doubling at similar intervals up to a total dose of 0.25 mg/kg. Side effects coincide with the onset of clinical effects and occur in up to a third of patients but are usually minor. These include flushing, dyspnea, chest pressure, nausea, headache, dizziness, transient bradycardia or heart block, and hypotension (seen rarely, from the vasodilatory properties). These side effects usually resolve rapidly without treatment, although many patients are intensely uncomfortable for a short period. Asystole can occur but is usually transient though unsettling to those observing. • Narrow-complex (QRS less than 0.12 second) tachycardias (regular and irregular) • Wide-complex (QRS 0.12 second or greater) tachycardias (regular and irregular) The 12-lead ECG is essential for evaluation of any patient with a suspected dysrhythmia. Use of a single ECG lead is often adequate for diagnosis, but multiple leads are often required to detect such things as the presence or absence of P waves (often best seen in inferior leads or V1-2; Fig. 79-7), the relationship between P waves and QRS complexes, prolongation of the QRS and QT interval, and evidence of ischemia or prior myocardial infarction (Box 79-4). For certain conditions, such as Brugada’s syndrome, the 12-lead ECG together with a history of syncope is diagnostic.
Dysrhythmias
Cardiac Cellular Electrophysiology
Anatomy and Conduction
Mechanisms of Dysrhythmia Formation
Classification of Antidysrhythmic Drugs
Class IC Agents
Class II Agents
Class III Agents
Amiodarone
Ibutilide
Dofetilide
Dronedarone
Class IV Agents
Miscellaneous Agents
Adenosine
Approach to Dysrhythmia Recognition and Management
Initial Assessment of Stable Patients

Full access? Get Clinical Tree


Dysrhythmias