110 Disorders of Water Balance
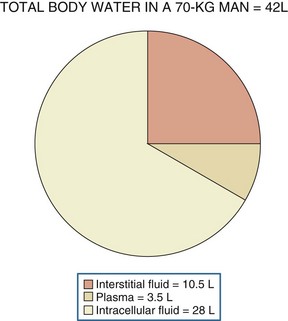
Water, the body’s most abundant constituent, accounts for approximately 50% of lean body mass in females and 60% of lean body mass in males. As shown in Figure 110-1, total body water is distributed between the intracellular compartment (two-thirds of total body water) and the extracellular compartment (one-third of total body water). The extracellular compartment is subdivided into the interstitial compartment (three-fourths of extracellular body water) and the plasma compartment (one-fourth of extracellular body water).1
The concentration of solutes in body fluids, as reflected in extracellular fluid by the serum sodium ion concentration, is tightly regulated between 138 and 142 mmol/L. This precise control is achieved by the maintenance of water balance; intake and losses are matched in a steady-state situation, despite marked fluctuations in daily solute and water intake. Water intake is determined primarily by thirst. Water excretion is controlled by the hypothalamic secretion of vasopressin (antidiuretic hormone [ADH]) and its target tissue, the renal collecting tubule. This allows for enormous flexibility because the kidney is able to dilute or concentrate urine (osmolality as low as 50 mOsm/kg H2O or as high as 1200 mOsm/kg H2O), depending on the body’s need to excrete or retain water, respectively. Under water-loading conditions, the kidney can excrete up to 20 to 25 L of urine a day. Likewise, the kidney has the ability to excrete as little as 0.5 L of urine per day (under conditions of water deprivation).1
 Control of Serum Sodium Concentration
Control of Serum Sodium Concentration
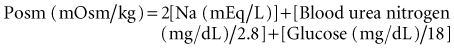
Under normal physiologic conditions, plasma osmolality is maintained between 280 and 290 mOsm/kg. Fluctuations in plasma osmolality outside this range are sensed by osmoreceptors in the hypothalamus, which is normally the primary determinant of the secretion of vasopressin, a cyclic octapeptide synthesized and secreted by supraoptic and paraventricular nuclei within the hypothalamus. The threshold for the osmotic release of vasopressin is 280 to 290 mOsm/kg, and the receptors are sensitive to changes in plasma osmolality of as little as 1% (Figure 110-2). The stimulus for vasopressin release is not limited to changes in osmolality. The primary nonosmotic stimulus for vasopressin secretion is decreased effective arterial blood volume, which can achieve a far greater rise in vasopressin levels than hyperosmolality can. Additional nonosmotic stimuli for vasopressin secretion include nausea, hypotension, and pain.
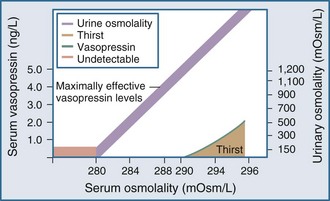
The primary site of action for vasopressin is within the principal cells of the renal collecting ducts. As illustrated in Figure 110-3, vasopressin binds to the V2 receptors on the basolateral membrane of these cells. Through a G protein–activated cascade, this results in increased insertion of a specific water (aquaporin 2) in the luminal membrane2 and renders the collecting tubule permeable to water.
Thirst also plays an important role in water balance. The most potent stimulus for thirst is hypertonicity; a change of 2% to 3% in plasma osmolality produces a strong desire to consume water. The threshold that triggers the sensation of thirst is higher than that for the release of vasopressin and usually occurs at a plasma osmolality of 290 to 295 mOsm/kg (see Figure 110-2). A decrease in effective arterial blood volume also stimulates thirst.
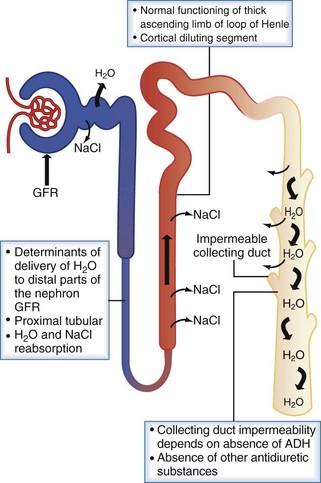
Protection against states of water excess is provided by the normally functioning renal diluting system. The three essential components of the diluting mechanism are depicted in Figure 110-4. First, because the major site of urine dilution is the water-impermeable ascending limb of the loop of Henle and the distal convoluted tubule, it is necessary to have normal delivery of tubular fluid to the distal nephron. Therefore, either a decreased glomerular filtration rate or increased proximal tubule fluid reabsorption limits the volume of dilute urine available for excretion. Second, the diluting segment of the nephron must be functioning normally. Thiazide diuretics, for example, impair the distal convoluted tubule’s ability to maximally dilute tubular fluid by blocking the thiazide-sensitive Na+/Cl− channel. Third, in order to excrete a dilute urine, vasopressin must be absent so that the collecting duct remains impermeable to water. With this diluting system intact, the kidney can handle a large load of free water (up to 1 L/h) without changes in serum sodium and thus serum osmolality.
An individual’s average daily solute load is approximately 600 mOsm. In states of low water intake, the kidney can concentrate the urine to 1200 mOsm/kg, therefore allowing for the excretion of as little as 0.5 L of urine per day. For this to occur, the renal concentrating mechanism must operate normally. The determinants of the renal concentrating mechanism are depicted in Figure 110-5. The water-impermeable thick ascending loop of Henle actively reabsorbs sodium chloride into the medullary interstitium while leaving water behind in the tubular fluid. The reabsorbed sodium increases the osmolality of the interstitium, which reaches its maximum at the papillary tip of the medulla. In the presence of vasopressin, water in the collecting duct is able to travel down its osmotic gradient and is reabsorbed. Once vasopressin is secreted, the collecting duct must be able to respond to it. Any disorder or pharmacologic agent that impairs the ability of vasopressin to act on the collecting ducts will incapacitate the renal concentrating mechanism and lead to dilute urine excretion.
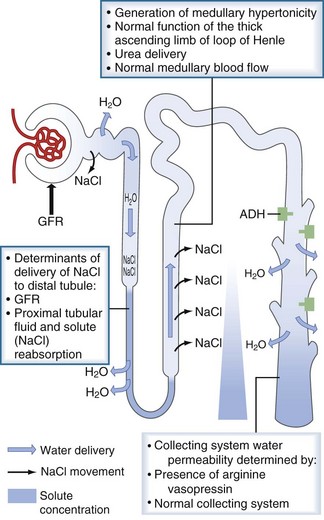
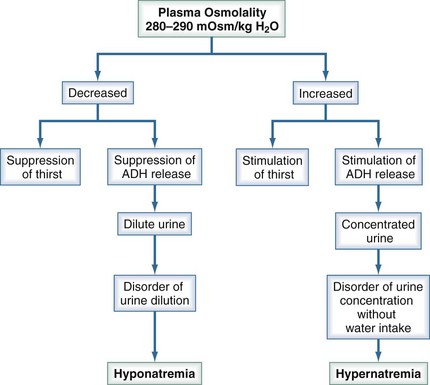
Figure 110-6 summarizes the mechanisms that maintain plasma tonicity and culminate in altered serum sodium values when impaired. These disorders arise whenever there is a disturbance in the body’s regulation of the relative amount of water to sodium. Hypernatremia results from a decrease in water relative to sodium (a water deficit state), and hyponatremia is caused by an increase in water relative to sodium (a water excess state).
 Hypernatremia
Hypernatremia
Hypernatremia is defined as a serum sodium concentration greater than 145 mEq/L. Its incidence in hospitalized patients ranges from 0.63% to 2.23%, with the elderly being more susceptible.3 Hypernatremia results in significant morbidity and mortality, ranging from 42% to 70% in adult patients. Acute elevations of serum sodium above 160 mEq/L are associated with a mortality rate of 75%, whereas mortality in chronic hypernatremia is 10%.
Hypernatremia develops whenever intake is less than the sum of extrarenal and renal water losses or, less commonly, when too much salt is introduced without adequate water intake. The primary defense mechanism against water depletion and hyperosmolarity is the renal concentrating capacity. However, even maximally concentrated urine does not prevent all water losses. Thirst also plays an important role in preventing water depletion. So long as water losses can be replaced, normal serum sodium concentration can be maintained. Most hypernatremic patients therefore have either an inability to obtain free water or an impaired thirst sensation. Both hypernatremia and hyponatremia can be assessed by the extracellular volume state: hypovolemic, isovolemic, or hypervolemic (see Figure 110-6).
Isovolemic Hypernatremia
Specific Isovolemic Hypernatremic Disorders
Central Diabetes Insipidus
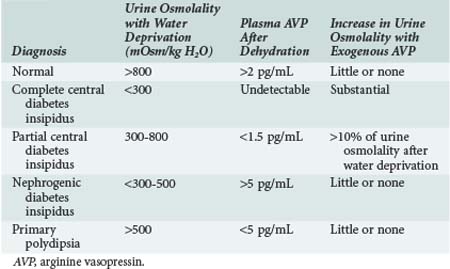
Central diabetes insipidus results from impaired secretion of vasopressin from the supraoptic and paraventricular nuclei of the hypothalamus. Known causes include congenital defects and infection, tumor, or trauma affecting the central nervous system (CNS); however, approximately 50% of cases are idiopathic (Box 110-1). Differentiating central diabetes insipidus, nephrogenic diabetes insipidus, and primary polydipsia can be a diagnostic challenge because all three present with polyuria and polydipsia. Several clinical features may assist in this effort. Central diabetes insipidus is often abrupt in onset with patients experiencing a constant need for water, whereas a compulsive water drinker often provides a more vague history of onset. Similarly, nocturia is common in patients with central diabetes insipidus but is unusual in compulsive water drinkers. The plasma osmolality is also a helpful measurement, with values above 295 mOsm/kg suggestive of central diabetes insipidus and values below 270 mOsm/kg favoring a diagnosis of compulsive water drinking. Distinguishing among the three entities is best accomplished by measuring vasopressin levels and monitoring the response to a water deprivation test followed by vasopressin administration (Table 110-1). Pituitary magnetic resonance imaging (MRI) can also be used to make the diagnosis of central diabetes insipidus. The T1-weighted images of a healthy posterior pituitary gland demonstrate a hyperintense signal, whereas this signal is absent in most patients with central diabetes insipidus (although it may be present in rare inherited forms of the condition).4
The treatment of central diabetes insipidus relies primarily on pharmacologic agents (Table 110-2). In the acute setting, aqueous vasopressin (Pitressin) is advantageous; its short duration of action makes complications such as water intoxication less likely. For a patient with chronic central diabetes insipidus, desmopressin acetate (DDAVP) is the agent of choice; it has a long half-life and can be administered intranasally (10-20 µg) every 12 to 24 hours. DDAVP does not have the strong vasoconstrictive properties of aqueous vasopressin, which must be used with caution in patients with coronary and peripheral vascular disease. In patients with partial diabetes insipidus, additional agents that increase the release of vasopressin (e.g., carbamazepine, chlorpropamide, clofibrate) can be used.
TABLE 110-2 Treatments for Diabetes Insipidus
| Type of Diabetes Insipidus | Drug | Dose |
|---|---|---|
| Complete central | DDAVP | 10-20 g intranasally every 12-24 h |
| Partial central | Aqueous vasopressin | 5-10 U subcutaneously every 4-6 h |
| Chlorpropamide | 250-500 mg/d | |
| Clofibrate | 500 mg 3-4 times daily | |
| Carbamazepine | 400-600 mg/d | |
| Nephrogenic | Thiazide diuretics | |
| NSAIDs | ||
| Amiloride (for lithium-related disease) | 5 mg/d | |
| Gestational | DDAVP | As for complete central |
DDAVP, desmopressin; NSAIDs, nonsteroidal antiinflammatory drugs.
Adapted from Lanese D, Teitelbaum I. Hypernatremia. In: Jacobson HR, Striker GE, Klahr S, editors. The principles and practice of nephrology. Philadelphia: CV Mosby; 1998.



Full access? Get Clinical Tree


