Chapter 82
Diabetic Ketoacidosis, Hyperglycemic Hyperosmolar State, and Alcoholic Ketoacidosis 
Diabetic Ketoacidosis
Definition
Diabetic ketoacidosis is defined by the presence of the following: hyperglycemia (blood glucose > 250 mg/dL), metabolic acidosis (arterial blood pH < 7.3 or bicarbonate < 16 mEq/L), and ketonemia (serum ketones ≥ 1:2 dilution) (Table 82.1). Although most patients with DKA have type 1 diabetes, DKA may also occur in patients with type 2 diabetes. African-American patients with type 2 diabetes appear to be particularly vulnerable, as evidenced by rising rates of DKA in this population. DKA remains a significant source of morbidity, accounting for as many as 9% of hospital admissions among patients with an established diagnosis of diabetes.
TABLE 82.1
Comparison of Diabetic Ketoacidosis, Hyperglycemic Hyperosmolar State, and Alcoholic Ketoacidosis
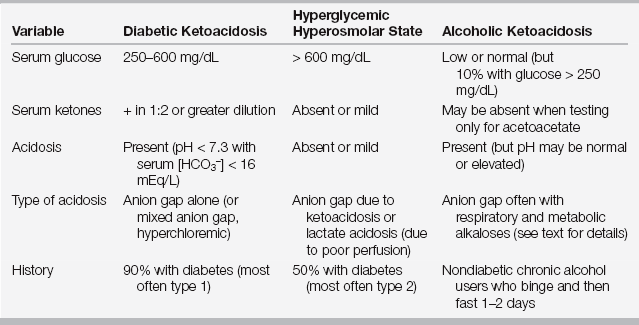
Pathogenesis
Diabetic ketoacidosis occurs when insulin deficiency and counter-regulatory hormone excess are present simultaneously. Glucagon is the most important counter-regulatory hormone, but other hormones in this category include cortisol, growth hormone, and epinephrine (Figure 82.1). As a result of this hormonal imbalance, there is increased glucose production via gluconeogenesis and glycogenolysis while glucose utilization in peripheral tissues is impaired. The hormonal milieu also favors the liberation of fatty acids that are subsequently oxidized to form ketones. The ketones, β-hydroxybutyrate and acetoacetate, account for the anion gap metabolic acidosis that is characteristic of DKA (Chapter 83). As glucose and ketones accumulate in the bloodstream, the reabsorptive threshold of the renal tubule is surpassed, resulting in glucosuria and ketonuria, respectively. The ensuing osmotic diuresis results in the loss of sodium, potassium, and water in the urine. Nonadherence with an insulin regimen frequently causes DKA, but other triggers must be sought. Infections, particularly urinary tract infections (UTIs), and pneumonia, myocardial ischemia, stroke, pancreatitis, drugs, and alcohol consumption commonly precipitate DKA.
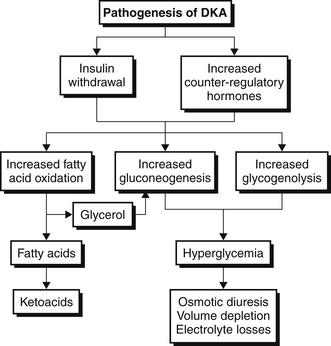
Evaluation
A thorough but rapid history is essential to determine the severity and duration of symptoms as well as to identify any precipitating events or comorbid illnesses. A schematic diagram for evaluation of the patient admitted to the ICU for DKA is shown in Figure 82.2. Presenting symptoms include polyuria and polydipsia that are a consequence of the hyperglycemia-induced osmotic diuresis. Gastrointestinal symptoms such as nausea, vomiting, and abdominal pain are common and are thought to be due to a combination of metabolic acidosis and ileus. Some patients may also report dyspnea due to the acidosis-induced increase in respiratory drive, which produces Kussmaul breathing/respirations.
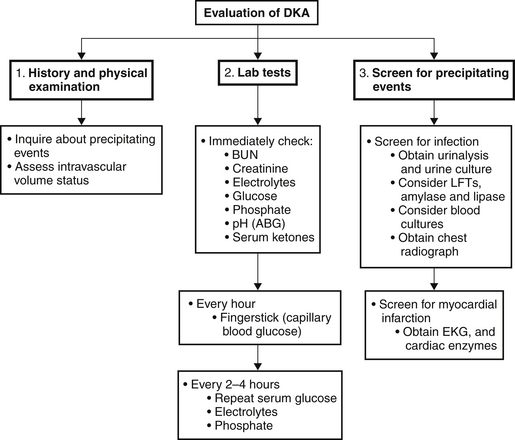
The acute metabolic acidosis of DKA typically is accompanied by an acute respiratory compensation. Although the acidosis is most commonly an anion gap acidosis, a simultaneous anion gap and nonanion gap acidosis may coexist; or rarely a pure nonanion gap acidosis may be found (see Chapter 83 on metabolic acidoses). The type of acidosis depends on the intravascular volume of the patient. If fluid intake is adequate and GFR preserved, the kidney excretes ketones, thus reducing accumulation of unmeasured anions that would contribute to the serum anion gap. The observed nonanion gap metabolic acidosis is due to ketonuria, which is akin to the loss of bicarbonate equivalents in the urine. On the other hand, patients who are volume depleted have less ketonuria and accumulate more serum ketones, resulting in an anion gap metabolic acidosis. Depending on the volume status, patients with DKA can fall anywhere along this spectrum.
Treatment
The treatment and management of DKA are outlined in Figure 82.3. The following treatment steps are the cornerstones of management: (1) volume resuscitation, (2) reversal of hyperglycemia, (3) inhibition of ketogenesis, and (4) repletion of electrolytes.
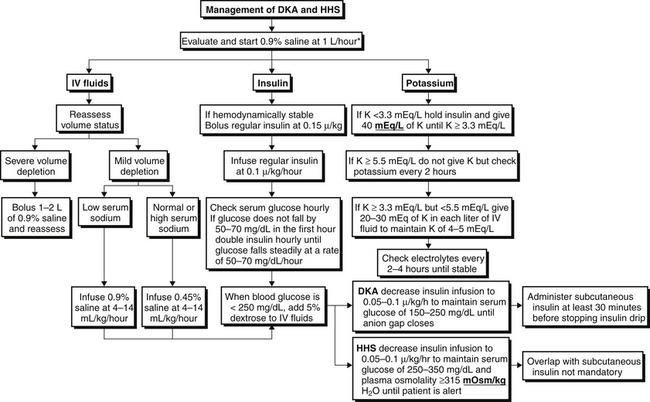
Volume Resuscitation
The average volume deficit is 3 to 4 L. At least 1 to 2 L of isotonic fluid (usually normal saline) should be administered rapidly. Patients with hemodynamic compromise should continue to receive volume resuscitation at a rate of 1 L/hour while their volume status is simultaneously monitored very closely. After adequate repletion of intravascular volume (as judged clinically), patients with a low serum sodium concentration should continue to receive isotonic fluid, but at a slower rate. Fluids are infused at 4 to 14 mL/kg/h, depending on the clinical assessment of volume status. On the other hand, if the serum sodium concentration is normal or elevated, the patient almost certainly has a concomitant free water deficit and the intravenous fluid therapy should be changed to 0.45% saline, also infused at a rate of 4 to 14 mL/kg/h. Serum osmolality should be corrected no faster than 3 mOsm per hour to avoid iatrogenic cerebral edema. The importance of frequent reassessments of volume status cannot be overemphasized, particularly for patients with a history of heart or renal failure.
Inhibition of Ketogenesis
Despite significant acidosis, bicarbonate therapy is rarely, if ever, necessary. Even with severe acidemia (pH 7.0 to 7.2), there is no clear evidence supporting the use of bicarbonate therapy. In fact, bicarbonate infusions can be detrimental because they may delay ketoacid metabolism, unmask hypokalemia, and cause intracellular acidosis. Increased bicarbonate drives PaCO2 production while simultaneously inhibiting respiratory drive, the result of which is intracellular acidosis. Bicarbonate therapy can be considered if the pH is < 7 but should be used judiciously.


Full access? Get Clinical Tree


