Key Clinical Questions
What are the most common cutaneous manifestations of gastrointestinal, renal, rheumatologic, and endocrinologic disease? What are rare but important cutaneous findings which should not be missed?
How does systemic amyloidosis manifest in the skin?
What key features are necessary to recognize nutritional deficiencies?
What is the systemic approach to purpura?
When should an inpatient dermatology consultation be obtained?
Introduction
This chapter will focus on the integument and its unique ability to exhibit disease that lies beneath. Though an all encompassing review of this subject is beyond the scope of this chapter, we will discuss the cutaneous manifestations of systemic disease that are commonly encountered during the daily practice of hospitalist medicine. A helpful approach to this subject is to consider whether a cutaneous lesion represents skin involvement by an internal condition, such as amyloidosis or cutaneous metastases, or a reactive process to underlying disease, such as neutrophilic dermatoses related to inflammatory bowel disease, or cutaneous vasculitis due to a drug hypersensitivity reaction. Additionally, cutaneous lesions can be characterized as specific to an underlying systemic process, or as a nonspecific manifestation that may occur in the setting of multiple disorders. Cutaneous findings may represent the sole clinical manifestation of a systemic disorder. Thus, a basic knowledge of these dermatologic conditions is essential not only for recognizing an underlying association, but also for determining when an inpatient dermatology consult may be useful in the diagnosis and management of an otherwise puzzling patient.
Cutaneous Manifestations of Gastrointestinal Disease
Disorders in which vascular lesions are found in both the skin and viscera may occasionally manifest with gastrointestinal (GI) hemorrhage. Though rare, this group of disorders should be considered when approaching a patient with a gastrointestinal bleed, particularly in the context of recurrent hemorrhage, familial history of hemorrhage, or multiple vascular lesions noted on the skin.
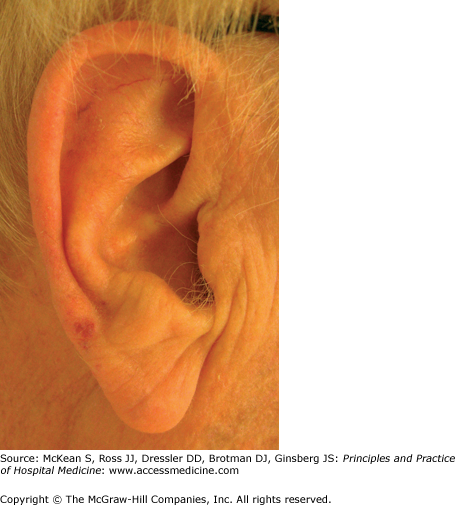
Telangiectasia in the setting of GI or pulmonary vascular lesions raises suspicion for hereditary hemorrhagic telangiectasia (HHT, also known as Osler-Weber-Rendu syndrome). This is a familial autosomal dominant or spontaneous disorder, characterized by cutaneous telangiectasias and visceral arteriovenous anomalies. Epistaxis in childhood is often the presenting sign, followed by the development of well defined 2–3 mm macular or papular telangiectasias on the face, lips, tongue, ears, and chest during late childhood to young adulthood (Figure 147-1). Telangiectasias represent dilations of the superficial capillary network, which typically blanch upon pressure. GI bleeding from mucosal telangiectasias occurs in about 20–40% of cases, with onset typically after the fourth decade. The stomach and duodenum are often sources of this potentially life-threatening bleeding. Additional visceral manifestations of HHT include hepatic or pulmonary arteriovenous fistulae, which may lead to shunting, dyspnea, hemoptysis, or systemic embolism and infection, and cerebral arteriovenous malformations, which may lead to intracranial hemorrhage.
Blue rubber bleb nevus syndrome (BRBNS) is a spontaneous or autosomal dominantly inherited disorder characterized by multiple venous malformations of the skin, GI tract, and other viscera. Cutaneous findings include multiple dark blue to violaceous compressible, rubbery skin lesions known as blebs, most commonly located on the trunk and upper extremities. Initially characterized in the literature as “hemangiomas,” the lesions associated with BRBNS are actually venous malformations that may appear macular, papular, or nodular. The classic “blue rubber nipple” demonstrates delicate crumpling when compressed with slow refill upon release. Notably, cutaneous lesions tend not to bleed but are characteristically tender due to the high content of vascular smooth muscle. Most blebs are present at birth or by 2 years of age, but may enlarge and become more prominent with age. It is important for hospitalists to be familiar with BRBNS, as potentially life-threatening GI bleeding is generally the most serious complication of BRBNS. Patients thought to have BRBNS should be screened with colonoscopy to evaluate for potential GI involvement.
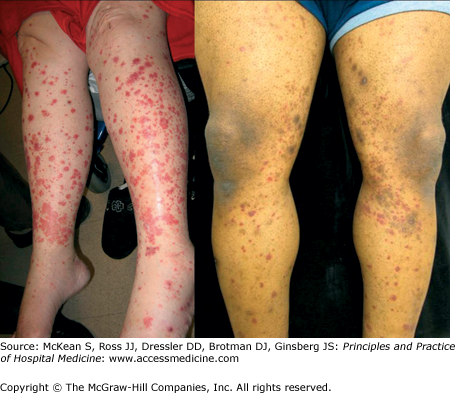
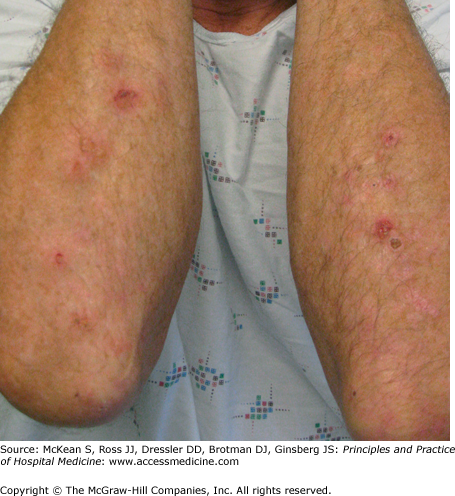
Henoch-Schönlein purpura (HSP) is a systemic vasculitis characterized by involvement of the skin, gastrointestinal tract, kidneys, and joints. Often considered a disease of childhood, HSP also occurs in adults. The major cutaneous manifestation of HSP is palpable purpura, the hallmark of small vessel vasculitis, presenting as 2–5 mm erythematous to violaceous papules that do not blanch with pressure (Figure 147-2). These lesions typically appear on the buttocks and lower extremities in HSP; lesions above the waist are anecdotally associated with an increased risk of renal involvement. Periumbilical purpura has also been noted to occur. Gastrointestinal signs and symptoms include abdominal pain, bleeding, and intussusception; these symptoms may precede the cutaneous findings. GI bleeding is often occult, and is suspected to occur in 40–60% of cases. If findings are limited solely to the skin, no treatment is necessary. If renal or GI involvement is present, more aggressive systemic immunosuppressive therapy may be necessary. Other systemic vasculitides, such as microscopic polyangiitis and polyarteritis nodosa, can also result in GI hemorrhage.
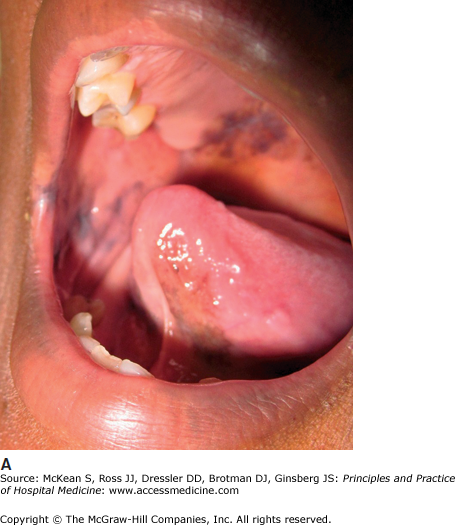
Kaposi sarcoma (KS) may occasionally present with gastrointestinal bleeding, though KS in general is much less common since the advent of highly active antiretroviral therapy for HIV. Cutaneous KS is a vascular neoplasm associated with human herpesvirus-8. The epidemic form of KS, which occurs in association with HIV, presents with violaceous patches, plaques, or nodules, most commonly on the chest, face, and oral cavity (Figure 147-3). Among patients with epidemic KS, involvement of the gastrointestinal tract is observed in 50–80% of patients with cutaneous disease, and more than 90% of patients with oral disease.
Pseudoxanthoma elasticum (PXE), a rare genetic disorder resulting in degeneration of elastic tissue, may affect the elastic media of blood vessels producing hemorrhage. Upper GI bleeding is more common than lower GI bleeding, and often presents in the third to fourth decade. Unlike the other disorders discussed in this section, the dermatologic manifestations of PXE do not typically include cutaneous vascular lesions. Instead, PXE manifests as yellow, cobblestoned papules that resemble “chicken skin” on the neck, axillae, antecubital fossae, and groin. Similar lesions or diffuse superficial erosions may be observed on mucosal surfaces during endoscopy. Ophthalmic signs of PXE include retinal neovascularization, producing angioid streaks. Other systemic manifestations include hypertension, premature vascular disease, cardiac disease, and cerebral hemorrhage. GI hemorrhage can be devastating in patients with PXE, and this hemorrhage may precede the classic dermatologic and ophthalmologic signs.
Malabsorption syndromes often produce nutritional deficiencies due to vitamin and mineral loss. An associated cutaneous eruption may suggest the correct diagnosis. Zinc deficiency may result from genetic, autosomal recessive abnormalities in zinc absorption, or be acquired from poor diet, geophagia (clay eating), chronic diarrhea, cystic fibrosis, total parenteral nutrition, renal disease, liver disease, alcoholism, or medications such as diuretics and sodium valproate. Zinc deficiency leads to a characteristic skin eruption known as acrodermatitis enteropathica. Eczematous, vesicular, or pustular skin lesions typically appear in an acral, perirectal, or perioral distribution. The diaper area is frequently involved. Alopecia, glossitis, and nail dystrophy may also be observed. Diagnosis is made via plasma zinc level, though alkaline phosphatase, a zinc-dependent enzyme, is also often low in this deficiency.
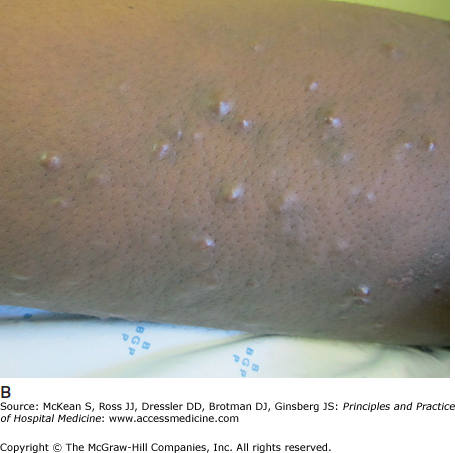
Dermatitis herpetiformis (DH) is characterized by a pruritic papulovesicular eruption on the extensor surfaces of the limbs, especially the forearms, elbows, and knees (Figure 147-4). The buttocks and posterior scalp are also often affected. As DH is intensely pruritic, patients often present only with excoriations, rather than well-defined vesicles, in the classic areas of involvement. DH is a cutaneous manifestation of gluten-sensitive enteropathy (celiac disease). Though more than 85% of DH patients demonstrate small bowel inflammation on biopsy, less than 10% have severe malabsorption. Patients may demonstrate an associated autoimmune disease or endocrinopathy, including thyroid disease, diabetes mellitus, or Addison’s disease. Treatment of DH includes gluten avoidance and antineutrophilic agents such as dapsone.
Ulcerative colitis (UC) and Crohn disease (CD) produce skin findings in as many as one-third of affected patients. Clinical manifestations range from specific lesions, such as fistulae formation and metastatic disease, to nonspecific reactive conditions. Many of these skin lesions may be manifestations of either UC or CD, though some findings are more often associated with a specific entity. Erythema nodosum and pyoderma gangrenosum will be discussed in detail. Other cutaneous manifestations of inflammatory bowel disease are listed in Table 147-1.
| Disease | Cutaneous Exam Findings | Association | Treatment |
|---|---|---|---|
| Nonspecific associations | |||
| Urticaria | Transient, edematous, pruritic wheals | UC, Crohn | Antihistamines |
| Aphthous ulcers | Painful oral erosions | UC, Crohn | Topical steroids |
| Small vessel vasculitis | Multiple, small (∼3–5 mm), nonblanchable red to purple macules and papules, known as “palpable purpura” | UC, Crohn | Systemic steroids if there is systemic involvement, otherwise supportive care |
| Erythema nodosum | Tender erythematous nodules on anterior shins, representing inflammation of the fat (panniculitis) | UC > Crohn | NSAIDs, bedrest, potassium iodide, systemic steroids, dapsone, colchicine |
Neutrophilic dermatoses
|
| UC >> Crohn |
|
| Cutaneous polyarteritis nodosa | Purple lacelike eruption on legs (livedo), with nodules or ulcerations. | Crohn | Systemic steroids, cytotoxic agents such as cyclophosphamide if systemic involvement |
| Specific associations | |||
| Fissures and fistula | Painful erosions and draining sinus tracts, most commonly in the perineum | Crohn > UC | Treat underlying Crohn; sinus tracts may require surgery |
| Cutaneous or metastatic Crohn | Cutaneous granulomas manifesting as erythematous plaques, nodules, and linear ulcerations, typically in inguinal crease | Crohn | Treat underlying Crohn |
| Mucosal Crohn | Cobblestoning and ulceration of oral mucosa appearing identical to bowel lesions | Crohn | Treat underlying Crohn |
| Rare associations | |||
| Erythema multiforme | Target lesions on extremities and trunk +/– ocular inflammation, hemorrhagic crusting on lips, oral/genital mucosal erosions | UC and Crohn (as well as infections and medications) | Supportive care, possible systemic steroids |
| Vitiligo | Depigmented macules on extremities, face, trunk | UC and Crohn (also thyroid disease) | Topical steroids, phototherapy |
| Epidermolysis bullosa acquisita (EBA) | Bullae and ulcerations with scarring over elbows and knees; may be peristomal (with differential diagnosis often including PG) | UC and Crohn | Treat underlying IBD, supportive care to bullae |
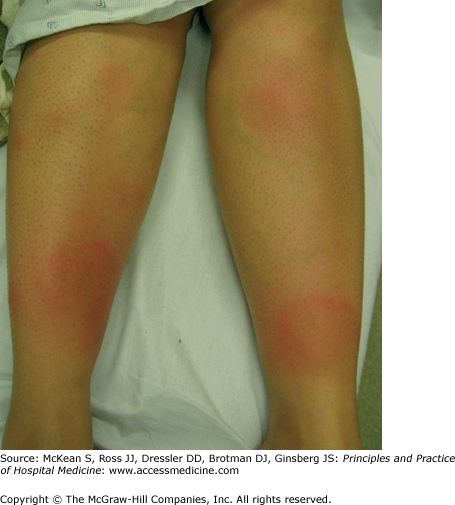
Erythema nodosum is the most common panniculitis or inflammatory disorder of fat. It presents with erythematous, tender nodules, characteristically on the shins (Figure 147-5). It is not specific for IBD. Erythema nodosum is often a hypersensitivity reaction to systemic disease or an antigenic trigger, although 30–50% of cases are idiopathic. Fever, malaise, and arthralgias may occur, and females are affected more commonly than males. It is typically self-limited and resolves within several weeks, with lesions appearing bruise-like later in the disease course. Ulceration usually does not occur. Erythema nodosum may precede the diagnosis of IBD or occur concomitantly with a disease flare. It occurs in 1–10% of patients with UC and in less than 2% of patients with CD. Common associations, evaluation, and treatment are provided in Table 147-2. The differential diagnosis includes other forms of panniculitis, such as connective tissue panniculitis, infectious panniculitis, pancreatic panniculitis, and erythema induratum or nodular vasculitis, which is often associated with tuberculosis.
| Select Causes | Diagnostic Considerations | Treatment |
|---|---|---|
|
|
|
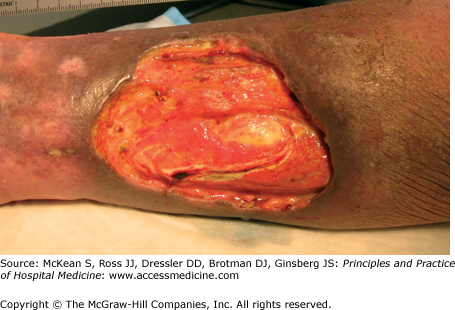
In 1930, Brunsting and colleagues characterized painful ulcers in several patients with underlying bowel disease. These skin lesions, which they called pyoderma gangrenosum (PG), were pustules that evolved into undermined ulcers with overhanging, gray borders and peripheral erythema (Figure 147-6). Although they were correct in identifying an association with bowel disease, the condition is not due to bacterial infection, as they had originally postulated. PG is now recognized as an inflammatory disorder sometimes associated with systemic diseases, including IBD, paraprotenemia, malignancy, and rheumatoid arthritis, although many cases are idiopathic. Though IBD is the single most common cause of PG, only 1–5% of patients with inflammatory bowel disease develop PG. In the setting of IBD, PG is usually associated with active colitis and may be an indication for surgery. PG typically presents on the leg, but lesions can present anywhere. Peristomal PG is also common in IBD patients. Disease associations and treatment for PG are summarized in Table 147-3.
| Select Systemic Associations | Treatment |
|---|---|
|
|
PG is a neutrophilic dermatosis, with diffuse skin infiltration by mature neutrophils. Other neutrophilic dermatoses include Behçet disease, neutrophilic eccrine hidradenitis, bowel bypass syndrome, rheumatoid neutrophilic dermatitis, and Sweet syndrome (acute febrile neutrophilic dermatosis). An important characteristic of each of these entities is the phenomenon of pathergy, meaning that pustular, neutrophil-rich skin lesions develop or worsen following intradermal trauma. The pathergy test used in the diagnosis of Behçet disease is based on this phenomenon. Evidence of pathergy may be found within sites of vaccinations, venipuncture, indwelling catheters, or recently performed procedures (Figure 147-7).
Figure 147-7
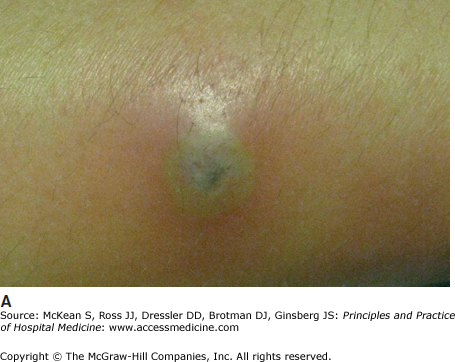
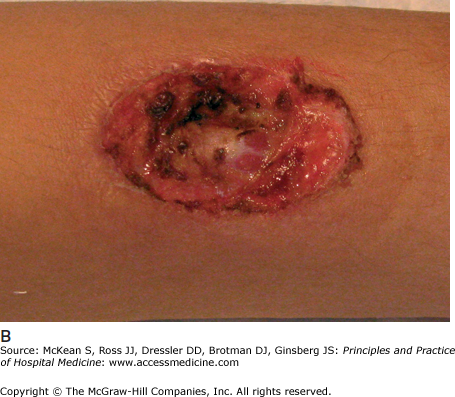
Demonstration of pathergy in a patient with pyoderma gangrenosum. A large pustule developed in this patient with ulcerative colitis (A), which was initially suspected to represent an infectious process; all cultures were negative. Following debridement of the lesion, an ulcer characteristic of pyoderma gangrenosum developed (B). This patient’s condition improved with systemic corticosteroids.
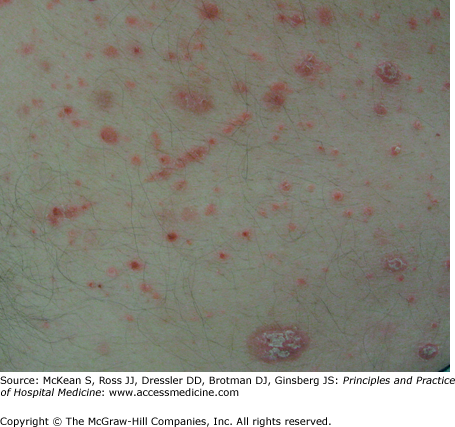
It is useful to differentiate pathergy from another term commonly used in dermatology known as the Koebner phenomenon (also called the isomorphic effect). First described in patients with psoriasis by Heinrich Koebner in the late nineteenth century, the Koebner phenomenon more generally refers to the development of lesions after minor traumatic insult. It occurs in up to 25% of patients with psoriasis, often resulting from mild trauma such as a sunburn or scratch (Figure 147-8). In addition to psoriasis, skin conditions that may demonstrate the Koebner phenomenon include lichen planus, vitiligo, and leukocytoclastic vasculitis. Unlike pathergy, koebnerization involves the development of lesions characteristic to an underlying disorder, and it is not a phenomenon limited to the neutrophilic dermatoses.
Cholestasis and cirrhosis have several cutaneous findings that are not specific to any particular etiology of liver disease. Diffuse pruritus and xerosis of the skin are common. Jaundice results from the binding of bilirubin to elastin, and may be observed initially in the conjunctivae, lingual frenulum, and on the soft palate, where elastic fibers are highly concentrated. Spider angiomata, with punctate central arterioles and branches towards the periphery (Figure 147-9A), are common in chronic liver disease, but may also be seen with oral contraceptive use, increasing age, alcohol use, and in chronic inflammatory conditions. Palmar erythema, affecting the thenar and hypothenar eminences, is another clinical sign of liver disease (Figure 147-9B). It is also seen in pregnancy, rheumatoid arthritis, lupus, thyrotoxicosis, diabetes, neoplasms, where it is thought to result from systemic release of angiogenic factors, and as an idiopathic condition. In liver disease, the pathogenesis of spider angiomata and palmar erythema is thought to involve increased levels of circulating estrogens.
Figure 147-9
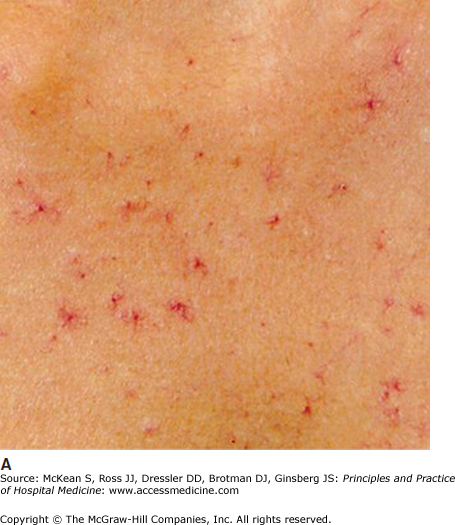
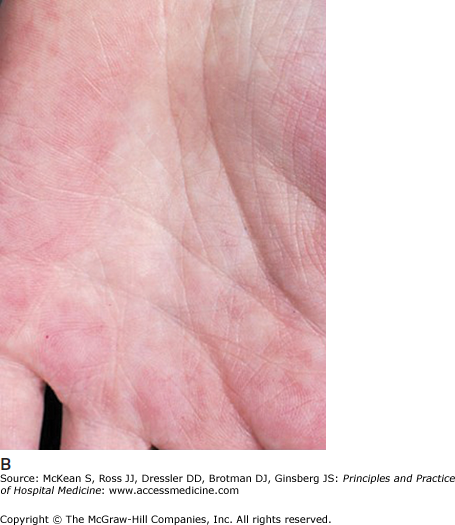
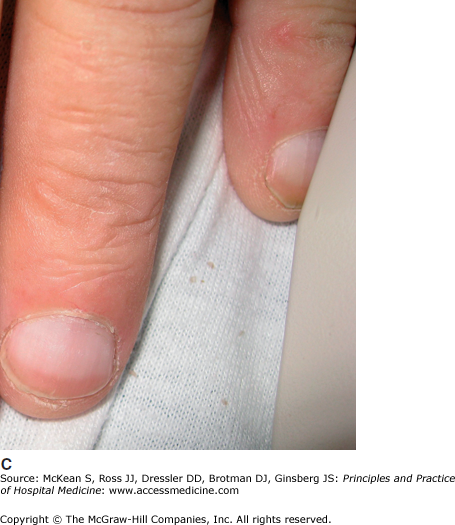
Cutaneous manifestations of liver disease. (A) Multiple spider angiomata. (Reproduced, with permission, from Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005. Fig. 151-9.) (B) Palmar erythema. (Reproduced, with permission, from Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005. Fig. 151-8). (C) Terry nails (Courtesy of Dr. Adam Lipworth).
Dupuytren contracture, a thickening of the palmar aponeurosis that may lead to fixed flexion of the fourth and fifth digit, is seen in some cirrhotic patients. It has also been associated with diabetes, anticonvulsant use, a positive family history, and possibly repetitive trauma. The nails are also commonly affected in liver disease. Terry nails, the classic nail finding in liver failure, demonstrate whitening of the majority of the nail plate, with retention of only a small band of pink nail plate along the distal portion of the nail (Figure 147-9C). Terry nails are also seen in renal disease and malnutrition. Chronic infection with hepatitis B virus (HBV) or hepatitis C virus (HCV) may be associated with these nonspecific findings of liver disease. Several skin disorders are also specifically associated with either HBV or HCV (Table 147-4).
| Hepatitis | Disease | Cutaneous Manifestations | Systemic Manifestations |
|---|---|---|---|
| HBV | Serum sickness reaction | Urticarial or morbilliform eruption | Proteinuria, hematuria, arthritis, hypocomplementemia |
| Polyarteritis nodosa | Purple lacelike eruption on legs (livedo), with nodules or ulcerations | Fever, abdominal pain, arthralgias, myalgias, mononeuritis multiplex, congestive heart failure, renal hypertension | |
| Papular acrodermatitis of childhood (Gianotti-Crosti Syndrome) | Skin-colored to red raised papules on extremities, buttocks and face, with sparing of the trunk | None | |
| HCV | Urticaria/urticarial vasculitis | Edematous wheals | Arthralgias, abdominal pain, nausea, vomiting, diarrhea, proteinuria, hematuria |
| Cryoglobulinemia | Red to violaceous raised papules on legs known as “palpable purpura,” purple lacelike eruption on legs (livedo), ulceration of skin | Arthralgias, neuropathy, membranoproliferative glomerulonephritis; rheumatoid factor (RF) positive in cases associated with HCV | |
| Lichen planus | Violaceous, pruritic, polygonal papules, often on extremities. Can demonstrate Koebner phenomenon. Oral involvement is most closely associated with HCV. | None | |
| Porphyria cutanea tarda (PCT) | Vesicles and bullae, predominantly on dorsal hands. Hyperpigmentation and fragility of skin, milia formation, hypertrichosis. | Porphyrins are typically detectable in the urine. Bright coral red fluorescence of patient’s urine with a Wood’s lamp allows for bedside diagnosis. | |
| Necrolytic acral erythema | Papulosquamous or vesicobullous eruption on hands and feet | May be associated with zinc deficiency | |
| Sarcoidosis (associated with interferon and ribavirin therapy for HCV) | Red to brown papules, plaques, rarely subcutaneous nodules | Hilar lymphadenopathy, respiratory or cardiac involvement, hepatomegaly, arthralgias/arthritis, nervous system involvement, ocular involvement (including lacrimal gland enlargement, uveitis) |
Cutaneous Manifestations of Chronic Renal Disease
The most common cutaneous complication of chronic renal failure is pruritus, which occurs in more than 50% of hemodialysis (HD) patients. Renal pruritus may be generalized, localized, sporadic, or continuous, and is exacerbated by HD. Though the precise etiology is unknown, it is suspected to involve an interplay between xerosis, neuropathy, mast cells, and possibly the HD membrane. Treatment strategies include emollients, narrowband phototherapy (to be used with caution in transplant patients or possible future transplant recipients due to an increased risk of nonmelanoma skin cancer, particularly squamous cell carcinoma), antihistamines, gabapentin, serotonin reuptake inhibitors, and opiate receptor antagonists.
Chronic renal failure is also associated with nail changes. Approximately 10% of patients with chronic renal failure demonstrate Lindsay nails, also known as half-and-half nails, in which the proximal portion of the nail appears white, while the distal half appears normal. In nephrotic syndrome, multiple white bands may appear parallel to the lunula of the nail; these bands are known as Muehrcke lines, and may also be seen with hypoalbuminemia due to liver disease or malnutrition.
Acquired perforating dermatosis is an uncommon, nonspecific manifestation of chronic renal disease (Figure 147-10A). These lesions, characterized by relatively small, dome-shaped papules with a hyperkeratotic central core or plug, occur in up to 10% of dialysis patients. Most patients have end-stage renal disease due to diabetic nephropathy and are undergoing HD. However, lesions may develop prior to dialysis, as well as in patients without renal disease who have diabetes, liver disease, or malignancy. The arms and trunk are predominantly affected, particularly on the extensor aspects. No intervention is necessary for these benign lesions, however, associated pruritus may be challenging to manage.
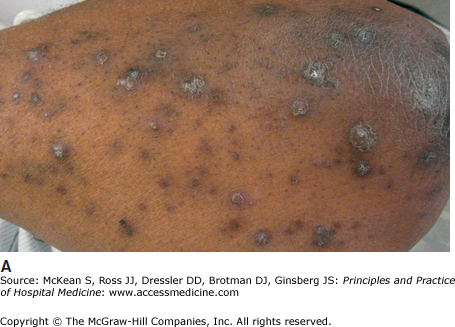
Nephrogenic systemic fibrosis (NSF), previously known as nephrogenic fibrosing dermopathy, is a progressive, fibrotic thickening of the skin, beginning on the lower extremities. As the disease progresses, joint contractures may occur, limiting mobility and ultimately disabling the patient. The pathogenesis of NSF seems to relate to oxidative tissue damage from free gadolinium ions in patients, typically those with renal failure, who undergo MRI contrast imaging studies; the risk is increased with progressive degrees of renal insufficiency, which lead to a longer tissue half-life of gadolinium, and is higher in patients who receive less-stable gadolinium chelates. Unfortunately, treatment options for NSF are limited. Patients should undergo physical therapy to limit loss of movement and avoid additional exposure to gadolinium. Pharmacologic therapy with immunosuppressives, thalidomide, and phototherapy has only a modest effect. Treatment with imatinib has shown some promising results in case series, though large randomized controlled trials have not been conducted.
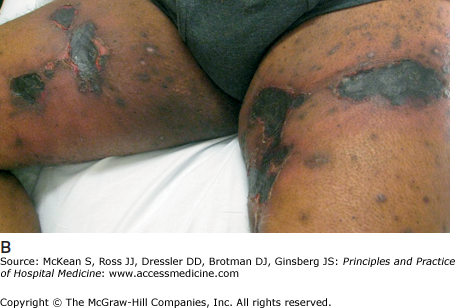
Calciphylaxis is a serious, potentially lethal condition that affects 1–4% of patients with end-stage renal disease (ESRD) on dialysis. Calciphylaxis can also occur in other settings, including coumadin use and primary hyperparathyroidism. It is a cutaneous vasculopathy characterized by arterial wall calcification and proliferation with subsequent fibrosis and thrombosis. The overlying skin develops progressive ischemia, with resultant severe pain and necrosis (Figure 147-10B). The pathophysiology is likely multifactorial, but may be related to prolonged hyperphosphatemia from secondary or tertiary hyperparathyroidism.
Clinically, calciphylaxis manifests as red to violaceous plaques that progress to nodules and deep, necrotic ulcers. Lesions may have an overlying eschar and appear stellate, or star-shaped, outlining the area of vascular compromise. There may be associated livedo racemosa or retiform purpura, a patchy, net-like pattern of violaceous erythema on the skin. There is a predilection for the thighs, abdomen, breasts, and other areas with a thick layer of adipose tissue. Women who are obese or who have type 2 diabetes are at increased risk.
Calciphylaxis can be rapidly progressive and potentially fatal, requiring prompt recognition and treatment. A skin biopsy should be performed for confirmation of diagnosis. Treatment options are limited (Table 147-5). Therapies aim to mitigate hyperparathyroidism and the attendant hypercalcemia and hyperphosphatemia. The severe pain associated with tissue ischemia must be addressed. Complications are common, including infection and poor wound healing due to chronic ischemia. Overall, the prognosis is poor, with a mortality rate of up to 85% following diagnosis. Proximal lesions have a higher mortality. The major cause of mortality in patients with calciphylaxis is sepsis from wound infections, indicating a crucial need for meticulous wound care.
Related posts:
 Strategies for Cost-Effective Care
Strategies for Cost-Effective Care
 Building, Growing, and Managing a Hospitalist Practice
Building, Growing, and Managing a Hospitalist Practice
 Designing a Hospitalist Compensation and Bonus Plan
The Face of Health Care Emerging Issues for Hospitalists
Designing a Hospitalist Compensation and Bonus Plan
The Face of Health Care Emerging Issues for Hospitalists
 Medical Malpractice
Preventing and Managing Adverse Patient Events: Patient Safety and the Hospitalist
Medical Malpractice
Preventing and Managing Adverse Patient Events: Patient Safety and the Hospitalist

Full access? Get Clinical Tree


