30 Critical Neuropathophysiology
 Elevated Intracranial Pressure
Elevated Intracranial Pressure
Physiology of Intracranial Pressure and Cerebral Blood Flow
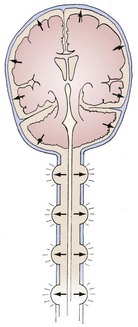
The brain, spinal cord, cerebrospinal fluid (CSF), and blood are encased in the protecting but noncompliant skull and vertebral canal, constituting a nearly incompressible system (Figure 30-1). In a totally incompressible system, pressure would rise linearly with increased volume. However, there is capacitance in the system, thought to be provided by the intervertebral spaces. Once this capacitance is exhausted, the ICP increases dramatically with increased intracranial volume.
Based on the following relationship:
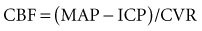
the concern is raised mathematically that increasing ICP is associated with decrements in CBF. However, the effect of increasing ICP on CBF is not straightforward, as mean arterial pressure (MAP) may increase with ICP elevations,1 and cerebral vascular resistance (CVR) adjusts with decreasing CPP (increasing cerebral vessel diameter) to maintain CBF until maximal vasodilatation occurs.2,3 This results in an increase in cerebral blood volume (CBV). This is thought to occur at a CPP less than 50 mm Hg, although considerable individual heterogeneity in this value exists with good reasons to believe that the lower limit of autoregulation (LLA) may be higher.4 Thus, increasing ICP initially is often associated with vasodilatation and/or increasing MAP to maintain CBF without a nutritive decrement.
Normal ICP is less than 10 mm Hg. ICP greater than 20 mm Hg is generally treated with ICP-reducing agents,5 but this is an epidemiologically derived action. Head trauma studies have indicated that patients with ICP over 20 mm Hg generally do poorly,5,6 although simply elevating ICP to above 20 mm Hg (in experimental animals) is not necessarily associated with decrements in CBF or permanent sequelae, provided the above-noted compensatory mechanisms occur,7,8 and venous ICP-related venous outflow obstruction with positive-feedback exacerbation of ICP does not occur.9
Nonetheless, increasing ICP due to mass lesions or obstruction of CSF outflow can exhaust compensatory mechanisms, with compromise of CBF. Initially, distal runoff of the cerebral circulation increases. As the process continues, the normally continuous (through systole and diastole) cerebral perfusion becomes discontinuous (systolic perfusion only) (Figure 30-2).10 Further compromise of CPP results in further oxygen extraction progressing to anaerobic metabolism, exacerbation of edema, and ultimately intracranial circulatory arrest.10 Thus, when ICP increases, early recognition is important to determine whether a deleterious sequence of events is starting.
Traditional notions of cerebral autoregulation, with CBF constant over a CPP range of approximately 50 to 150 mm Hg, has not gone without challenge.4 Drummond argues that this common notion derives from a figure in a review article by Lassen,11 which itself was an estimate based on data published by McCall in 195312 from pregnant volunteers undergoing blood pressure (BP) alteration with hydralazine and Veratrum viride. Despite the use of potentially cerebral vasoactive drugs, these observations remained unconfirmed in humans. Drummond suggests that most human data published since 1953 support an LLA of 70 mm Hg, with one investigator suggesting the onset of cerebral ischemia symptoms in normal humans to arise at a MAP of 55 mm Hg.13 Moreover, his closer review of published data suggests large interindividual variation in LLA. Drummond suggests that the only safe approach to an individual patient is to assume that no less than 75% of his/her resting MAP should be assumed to be the LLA. Symptoms of cerebral hypoperfusion tend to arise when MAP falls to about 50% of the resting value. These assertions of the need to individualize are increasingly being supported in the context of head injury with recent studies of the use of dynamic autoregulation assessment to determine optimal BP for a given patient.6,14–16
It is also of interest that LLA, based on CPP (MAP − ICP), may vary with ICP and with jugular venous pressure. McPherson et al., in a canine model, noted LLA was higher with elevated jugular venous pressure. This may, however, actually reflect the lack of knowledge regarding the proper definition for CPP, and that it may vary depending on the influence of the venous Starling resistor.8,17 Brady et al.,18 in an atraumatic immature piglet model of intracranial hypertension, found that LLA had a positive correlation with ICP. That is, LLA CPP was higher with higher levels of ICP. They suggest the possibility that compensating for an increase in ICP with an equivalent increase in arterial blood pressure (ABP) may not be sufficient to prevent a decrement in CBF and cerebral ischemia. Further studies in adults will be needed. Nonetheless, Brady et al.18 point out that Cremer et al.19 observed an LLA elevation in adult trauma patients with intracranial hypertension. The overall suggestion is that there is a need for individualized dynamic autoregulation assessment to determine each patient’s optimal CPP.6,14–16 Indeed, this may be only one component of a battery of multimodal monitoring, so-called integrative neuromonitoring, that is increasingly being advocated.20
Another approach to characterizing cerebral autoregulation has been espoused by Dewey et al.,21 Early et al.,22 and Burton et al.23 Using observations in pacemaker-dependent dogs and a beat-to-beat measure of brain blood flow, they observed that abrupt cessation of cardiac activity produced zero CBF well above the generally accepted LLA. Indeed, they reported that this critical closing pressure varied with the resting MAP, generally being about 40 to 50 mm Hg below MAP. They concluded that the normal cerebral circulation assumes a tonic state of contraction that varies with MAP and ICP, more tone at higher BP or lower ICP, less at lower BP or higher ICP, such that the true dynamic cerebral perfusion pressure is MAP − CCP, with CBF = (MAP − CCP)/CVR. Burton’s model can be use to describe CCP as: CCP = ICP + tension of arterial walls.21 The CCP is presumed to be altered by various drugs and disease states to thereby produce variations in CBF, despite otherwise unchanged traditional CPP (MAP − ICP). Thus the true definition and measurement of CPP may be a good deal more dynamic and complex than is understood at this time.
CCP was further studied more recently by Czosnyka et al.24 in humans with traumatic brain injury (TBI). If autoregulation is relatively intact, CCP-ICP remains high, but with injury sufficient to produce dysautoregulation, CCP-ICP decreases, indicating decreased tension in arterial walls.
Contributors to Intracranial Hypertension
Brain
The brain normally occupies about 80% of the contents of the skull, but its volume can be increased by edema. There are two types of edema, cytotoxic and vasogenic, referring to swelling produced by cellular or vascular processes, respectively.25 Any edema can increase ICP. It can be heterogeneously distributed such that pressure gradients occur, leading to a variety of herniation syndromes.
Cerebrospinal Fluid
CSF is generated in the choroid plexus and absorbed in the arachnoid villi. An equilibrium normally exists between production and absorption. Disruption of this equilibrium can lead to increased ICP with hydrocephalus, the condition wherein there is an excess of fluid in all or part of the CSF in the brain. Hydrocephalus is generally categorized as communicating or noncommunicating. In communicating hydrocephalus, the CSF circulation between the site of CSF production and absorption is intact. However, abnormally decreased absorption or increased production results in increased CSF accumulation. In noncommunicating hydrocephalus, the pathways are blocked such that CSF cannot circulate to the convexity of the brain to be absorbed. This results in accumulation of CSF in the ventricles, producing distension.26
Blood
Another mechanism of increased CBV occurs with obstruction of venous outflow. This results in brain engorgement and edema and CBV-mediated increased ICP, but without increased CBF27; this too is discussed in more detail later.
Venous Pathology
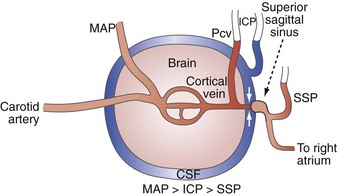
Venous pathology also plays a role in the genesis and propagation of intracranial hypertension. Blood coursing through the brain runs through arteries, capillaries, veins, sagittal and other dural sinuses, and then on to the internal jugular and other extracranial veins. In the context of a closed intracranial space, the relationship of these vessels to the tissue and CSF surrounding them becomes important. Notably, several investigators, in laboratory preparations, have observed a distinct drop off in intraluminal pressure in going from cerebral cortical veins to the sagittal sinus. This is most evident when ICP is elevated and indicates the presence of a vascular waterfall at a point just proximal to the sagittal sinus as the extraluminal high-pressure CSF acts to impede flow from cortical veins to the sagittal sinus.8,9,28,29 In fact, Nemoto9 and Nakagawa et al.28 have further observed that the cerebral venous pressure tends to be consistently higher than the ICP. The implications of these observations are that elevated ICP begets increased cerebral venous pressure. The increased cerebral venous pressure promotes and exacerbates brain edema, which may have been the initial cause of the intracranial hypertension. This then leads to a positive-feedback cycle wherein increased ICP increases cerebral venous pressure which then increases ICP.9,30 Thus any other factors that may promote brain edema or otherwise increase ICP in this tenuous situation (e.g., high extraventricular drain, systemic hypertension,31 hypoosmolarity) may initiate such a positive-feedback process.
Types of Intracranial Hypertension
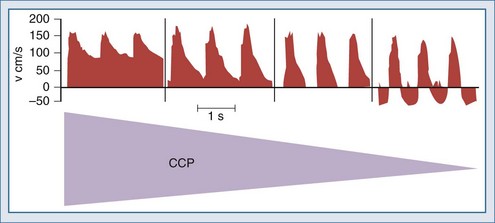
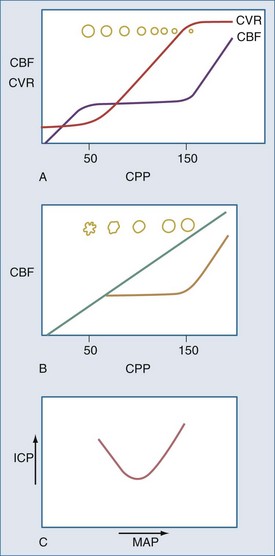
There are two types of intracranial hypertension, categorized according to CBF as hyperemic or oligemic (Figure 30-3).6 In the normal state, increases in CBF are not associated with increased ICP, because capacitive mechanisms compensate for the CBV-mediated increased intracranial volume. However, in the situation of disturbed intracranial compliance, small increases in intracranial volume produce significant increases in ICP.2,3
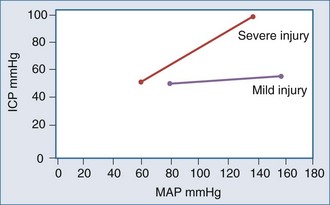
For many years it has been known that abrupt noxious stimuli briefly increase ICP in the setting of decreased intracranial compliance. Recent studies have revealed that such situations are associated with hyperemia, strongly suggesting that brief hyperemic intracranial hypertension is not a dangerous situation.32 However, it is reasonable to be concerned about such hyperemia for four reasons. First, elevated ICP due to hyperemia in one portion of the brain may increase ICP to compromise CBF in other areas of the brain in which CBF is marginal. Secondly, increased pressure in one area of the brain may produce gradients that might lead to a herniation syndrome. Thirdly, there is theoretical concern that inappropriate hyperemia predisposes the brain to worsened edema or hemorrhage as occurs with hyperperfusion syndromes. And fourthly, there is increasing evidence that increased ICP obstructs venous outflow to further exacerbate brain edema in a positive-feedback manner.9,30 Thus hyperemic intracranial hypertension has a theoretical potential to be deleterious, although this has yet to be demonstrated in a systematic fashion. For brief periods, as may occur during intubation or other limited exposure to noxious stimuli, it is suggested (but not proven) that it may not be problematic.33 An example of this conundrum is illustrated in Figure 30-4.
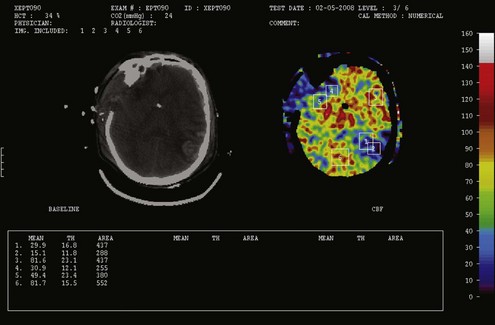
In contrast, oligemic intracranial hypertension is associated with compromised cerebral perfusion and is clearly deleterious.6,10 This is supported by the high mortality rate observed in head trauma patients in whom ICP rises due to brain edema with decrements in CBF.10,34 Transcranial Doppler echography and CBF studies on these patients have demonstrated that CBF is low and perfusion is discontinuous during the cardiac cycle (see Figure 30-2).10,35 Moreover, jugular venous bulb data indicate that oxygen extraction is markedly increased, suggesting loss of reserve with occurrence of anaerobic metabolism.35 In this setting, noxious stimuli can further increase the ICP, producing the situation of hyperemic added to oligemic intracranial hypertension. Presumably, in this setting the hyperemic rise in ICP acts to further reduce regional CBF in compromised areas with brain edema and may contribute to vasogenic edema.
Blood Pressure Effects on Intracranial Pressure: Plateau Waves
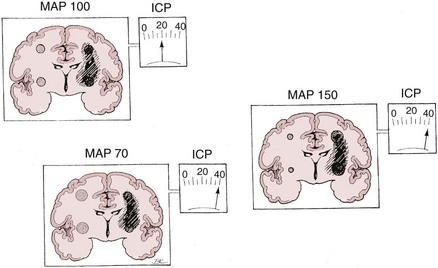
Lundberg, in a pioneering 1960 study,34 monitored ICP in hundreds of patients, identifying characteristic pressure waves. One category of these waves has been identified as plateau waves, which are known to be associated with increased CBV (Figure 30-5).2 Such waves occur when the ICP abruptly increases to systemic BP levels for about 15 to 30 minutes, occasionally accompanied by neurologic deterioration. Rosner3 synthesized the data and convincingly suggests that intracranial blood volume dysautoregulation is responsible for plateau waves. He induced mild head trauma in cats and subsequently intensively monitored the animals after the insult. With normal fluctuations in BP, while in the normal range, he observed that mild BP decrements to a mean of approximately 70 to 80 mm Hg preceded the development of plateau waves (Figure 30-6). Cerebral blood volume in normally autoregulating brain tissue increases with decreasing BP. However, the increase in CBV is nonlinear. There is an exponential increase in CBV as CPP decreases to levels of 80 mm Hg and below (Figure 30-7).3 A small decrease in BP, although in the normotensive range, produces exponential increases in CBV in a setting of abnormal intracranial compliance with the ICP at the elbow of the ICP-intracranial volume curve. Thus a small decrease in BP introduces an exponential CBV change upon an exponential ICP relationship such that ICP will increase abruptly and to a significant extent. Cremer et al.’s observations of ICP increases with deliberate ABP decreases in TBI patients provide further support that these concepts are relevant to clinical practice.19
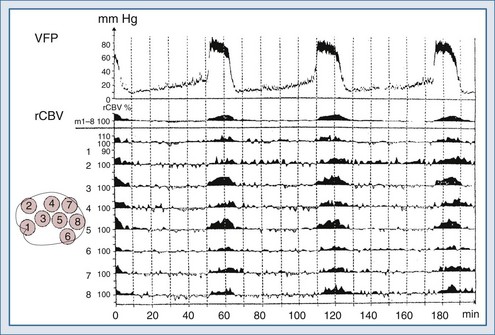
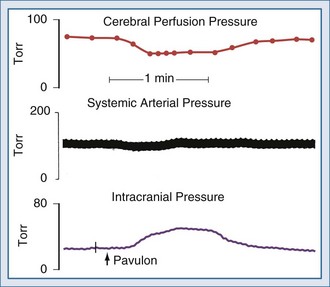
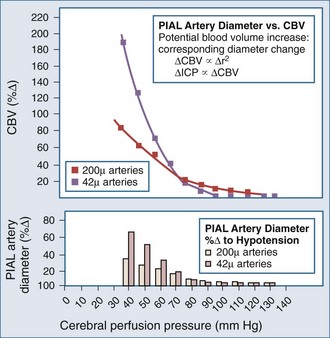
Figure 30-7 Cerebral vasodilatation occurs exponentially as cerebral perfusion pressure is reduced.
(From Rosner MJ, Becker DP. The etiology of plateau waves: a theoretical model and experimental observations. In: Ishii S, Nagai H, Brock M, editors. Intracranial Pressure V. New York: Springer-Verlag; 1983:301.)
Conversely, hypertension can also increase ICP, with animal models showing increased brain water with dopamine-induced increased blood pressure.31,36 Typically, within the normal autoregulatory range, changes in BP have no effect on ICP. However, with brain injury and associated vasoparalysis, BP increases mechanically to produce cerebral vasodilatation, increasing ICP (Figure 30-8).6,15,16,37–40
It appears that both increasing and decreasing BP can increase ICP, suggesting the presence of a CPP optimum for ICP—based on Rosner’s observations,3,6,41,42 probably about 80 to 100 mm Hg, although this has not been definitively determined experimentally (Figures 30-9 and 30-10). An alternate view that lower BP should be employed has been argued as the so-called Lund approach by Grände et al.,30 with much of its rationale based on the earlier discussion of the role of venous obstruction in intracranial hypertension. Indeed, recent studies are increasingly supporting the notion that the CPP optimum is highly variable and should be individually determined with emerging technologies.6,15,16
Blood Pressure, Brain Injury, and Intracranial Hypertension—Beyond Plateau Waves
Recent advances in transcranial Doppler (TCD) ultrasonography have allowed insights into dynamic, nearly instantaneous assessment of cerebral autoregulation in critically ill patients. Such technology has permitted the aforementioned observations of CCP in head-injured humans24 and the report of apparent diminution in arterial wall tension in patients with cerebral dysautoregulation. Moreover, Czosnyka et al.14 observed in TBI patients a U-shaped curvilinear relationship in flow velocity versus ABP, with worse autoregulation with ABP lower than 75 mm Hg and ABP higher than 125 mm Hg. They also noted increasing ABP to also increase ICP, further indicating a marker of dysautoregulation, the so-called PRx6,15,16 (see following paragraph). Dynamic time domain analysis of cerebrovascular autoregulation using near-infrared spectroscopy (NIRS) or TCD is a current topic of investigation with promising reports of potential efficacious and valid bedside use.15,39,43–45
The ICP pressure-reactivity index (PRx) is a more recently described quantitation of the earlier description of abnormal dynamic correlation of ICP changes with ABP changes and is another means to dynamically evaluate autoregulation,6,15,16,40 with reports indicating that PRx correlates well with other autoregulation indices.6,16,38,46 Steiner et al.15 reported on the use of PRx monitoring in TBI patients to determine the optimal CPP. Patients with better autoregulation as defined by PRx had better outcomes. Moreover, patients with dysautoregulation related to higher ABP with corresponding ICP elevation also had worse outcomes, suggesting that autoregulation monitoring to ensure adherence to an individual’s optimal CPP may be an outcome-altering intensive care unit (ICU) measure. Zweifel et al.6 report congruent observations. Notably, PRx, as with TCD-based autoregulation studies, also appears to undergo a U-shaped curvilinear relationship with variations in CPP, with it being abnormally high (i.e., ICP varies with ABP) at low (ischemic) and high (hyperemic) CPP in TBI patients. Further complementing this are observations of abnormally high oxygen extraction fraction (OEF) and low OEF at these respective ABP extremes. This is underscored by several reports of a significant ischemic burden in TBI patients,47–50 suggesting a delicate balance between hypotension-associated hypoperfusion and hypertension-associated edema exacerbation, both of which will worsen regional ischemia. Taken altogether, these autoregulation studies introduce the hypothesis that there is an individualized ABP optimum in TBI patients6 that should be a therapeutic goal.
Positive End-Expiratory Pressure and Intracranial Hypertension
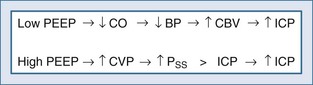
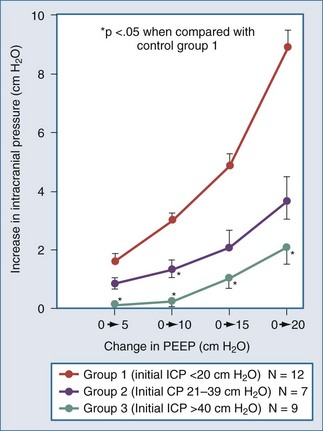
Positive end-expiratory pressure (PEEP) can increase ICP in two ways. The first is through impedance of venous return, increasing cerebral venous pressure and ICP. The second is through decreased BP and reflex increase of CBV, increasing ICP (Figure 30-11). The latter is likely the most common mechanism. Huseby’s data51 suggest that cerebral venous effects only occur with very high PEEP, a notion theoretically supported by the earlier discussion on the role of the veins in autoregulation of CBF and genesis of intracranial hypertension.
Shapiro and colleagues52 demonstrated increases in ICP in head-injured humans during intracranial hypertension with application of PEEP (Figure 30-12). Examination of their data suggests that the most profound decreases in CPP occurred in patients with PEEP-induced decrements in MAP. This is consistent with the view put forth by Rosner3 that decreases in BP increase CBV and ICP. Aidinis and colleagues,53 in studies on cats, confirmed these observations in a more controlled setting. In addition, they assessed the role of pulmonary compliance, finding that decreased pulmonary compliance induced by oleic acid injections results in less effect of PEEP to increase ICP. In situations in which PEEP is likely to be needed, with decrements in pulmonary compliance, such observations indicate that any adverse effects on ICP are less likely to be manifest. This may be related to observations that hemodynamic effects of PEEP are less apparent with noncompliant lungs,53,54 such that hypotensive-mediated increases in CBV do not occur.
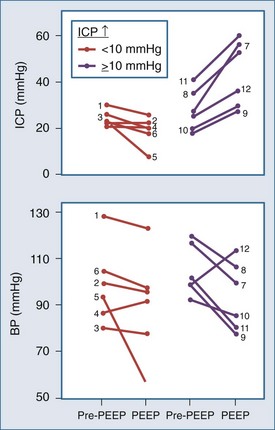
The intuitive notion that PEEP increases cerebral venous pressure to increase ICP is not as straightforward as it initially may seem. For PEEP to increase cerebral venous pressure to levels that will increase ICP, the cerebral venous pressure must at least equal the ICP, which affects the Starling resistor just proximal to the sagittal sinus.28 Thus the higher the ICP, the higher PEEP must be to have such a direct hydraulic effect on ICP. This concept was nicely proved by Huseby and colleagues51 in dog studies in which PEEP was increased progressively, with different starting levels of ICP (Figure 30-13). It is important to note that they prevented PEEP-induced decrements in BP, thus avoiding any reflex increases in CBV. They suggested a hydraulic model to better conceptualize this (Figure 30-14). For example, if all of a 10 cm H2O PEEP application were transmitted to the cerebral vasculature—which is unlikely given the decreased pulmonary compliance associated with the need for such PEEP—ICP will only be affected if it is less than 10 cm H2O (7.7 mm Hg), increasing to a level no higher than the applied PEEP. This presupposes no PEEP-induced arterial pressure decrement.
Antihypertensive Therapy Effects on Intracranial Pressure
Intracranial pressure can also be influenced by antihypertensive drugs. In general, vasodilator drugs such as nitroprusside,55,56 nitroglycerin,57 and nifedipine58 can be expected to increase ICP. Conversely, nonvasodilator antihypertensive drugs, generally sympatholytic drugs such as trimethaphan or beta-adrenergic blocking drugs such as esmolol or labetalol,59 can be expected to have little or no effect on ICP. These observations suggest that the rise in ICP due to vasodilators is caused by increased CBF with an attendant increase in CBV. Also, decreases in BP, as an indirect consequence, may produce vasodilation in autoregulation brain areas, with increased CBV and ICP as discussed earlier for plateau wave physiology. The increase in ICP by these direct and indirect mechanisms thus does not threaten ischemia directly, although herniation and hyperperfusion syndromes and the aforementioned issues with venous outflow obstruction may occur and might be problematic. There has been a report of neurologic deterioration with nitroprusside use despite no change in BP.56 Another consideration in the use of vasodilators is the propensity to reflexively increase endogenous plasma catecholamine concentrations.60 Such increases in plasma catecholamines may be deleterious to the marginally perfused injured brain.61–63



Full access? Get Clinical Tree


