Chapter 92 Congenital Immunodeficiencies
The discovery of antimicrobials in the middle of the last century allowed for the description of patients with defects of immune function. These classic descriptions recognized the primary immunodeficiency diseases (PIDs) in association with recurrent or persistent infection, infections with unusual organisms (opportunistic pathogens), or unusually severe presentations of infectious diseases with pathogens of low virulence. These patients became the experiments of nature that established the immune system as essential for host defense.1 The subsequent observation that many of these patients succumb to malignancy or autoimmune diseases further underscores the role that the immune system plays in the pathogenesis of these disease states.
Taken together, PIDs are a group of more than 150 different heterogeneous disorders of immune function in which more than 100 different defective genes have been recognized (Figure 92-1).2 The estimated overall prevalence varies, but is generally accepted to be close to 1:10,000. This suggests that there may be approximately 400 new cases of primary immunodeficiency in infants born in the United States each year (4.0 million live births).3 Early and prompt diagnosis is essential because curable and life-saving therapies are available. If a molecular diagnosis is established, prenatal diagnosis can be offered to parents and family members.
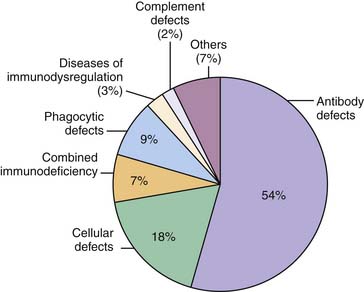
Figure 92–1 Distribution of the major primary immunodeficiency disorders.
(Data from The Jeffrey Modell Foundation Survey, 2009.)
Although there may not be a particular physical phenotype to encompass all these disorders because most children with PID appear normal, some physical findings that should make the clinician think about a congenital immunodeficiency include failure to gain weight, absent tonsils, persistent thrush, difficulties with healing, rashes and eczema that do not resolve with conventional therapies, severe warts, gum disease, cutaneous granulomas, unexplained hepatosplenomegaly, telangiectasias in eyes and ears, ataxia, dwarfism, cartilage abnormalities, and oculocutaneous albinism (Box 92-1).
There are particular syndromes associated with defects in immune function. These include, among others, DiGeorge syndrome, defects of DNA repair (ataxia/telangiectasia, Nijmegen breakage syndrome, Bloom syndrome), immuno-osseous dysplasias (cartilage/hair hypoplasia and Schimke syndrome), Hoyeraal-Hreidarsson syndrome, immunodeficiencies with associated defects in pigmentation (Chediak-Higashi syndrome, Griscelli syndrome, Hermansky-Pudlak syndrome), and those with associated phagocytic defects (Kostmann syndrome, Papillon-Lefevre syndrome, and Shwachman-Diamond syndrome).3
The Immune System and the Classification of the Primary Immunodeficiency Diseases
The human immune system has evolved to protect the individual from infectious microbes. It does this by using a complex interactive network of cells, proteins, and organs. This response is both innate and adaptive based on unique characteristics. The innate response to a pathogen occurs immediately (within hours), lacks clonal specificity for a particular pathogen, and does not confer long-lasting protection—that is, immunologic memory. Both cellular and humoral factors constitute its major components. These include (1) antimicrobial products and physical barriers such as the skin and mucosal surfaces; (2) receptors for pathogen molecules, including the family of Toll-like receptors (TLRs); (3) the phagocytic cells (neutrophils and macrophages); (4) dendritic cells; (5) the complement system; and (6) natural killer (NK) cells.4
The effector cells of the adaptive immune response are T and B lymphocytes. These cells derive from a hematopoietic stem cell (HSC) precursor. It is generally accepted that HSCs differentiate into a common lymphoid progenitor (CLP) that gives rise to T, B, and NK cells. Lymphopoiesis is tightly regulated and leads to the expression of a functional antigen receptor on the surface of the lymphocyte. For the B cell, it is the immunoglobulin molecule; for the T cell, it is the TCR complex. Cellular microenvironment, growth factors, cytokines, and chemokines, along with silencing or activating certain genes at different stages of lineage commitment, are some of the multiple factors contributing to a successful and mature lymphocyte. Although mouse lymphoid development has been well elucidated, human lymphoid development has been fragmented; our knowledge is patched together from in vitro analysis of bone marrow–derived cells, patients, and comparisons to the mouse models.4
Patients with PID have taught us that despite the apparent redundancy of the system, quantitative and qualitative defects in individual components and/or pathways result in abnormal function and susceptibility to particular infections.1
Historically, defects of cellular, humoral, phagocytic function, and complement defects constituted the classic classification of PID (see Figure 92-1). As understanding of the immune system has evolved, PIDs can also be considered as defects that involve either innate or adaptive immune responses. In the 1970s, the World Health Organization sponsored a committee of experts in the field of PID to classify and define these disorders. Since then, the International Union of Immunological Societies Expert Committee on Primary Immunodeficiencies has met regularly to update the classification of PID. The most recent publication was published in the Journal of Allergy and Clinical Immunology in December 2009.2 In this review, PIDs are classified as being (1) combined T- and B-cell immunodeficiency, (2) predominately antibody deficiencies, (3) well-defined immunodeficiency syndromes, (4) diseases of immune dysregulation, (5) predominately phagocytic defects, (6) defects of innate immunity, (7) autoinflammatory disorders, and (8) complement deficiencies. As new clinical phenotypes are identified and the genetics of these disorders are unraveled, future classifications will reflect advances in the understanding of disease pathogenesis.5
Laboratory Diagnosis of Congenital Immunodeficiencies
Most patients admitted to the ICU have many laboratory tests performed. It is important to recognize laboratory results that should not be arbitrarily dismissed as attributable to the severity of the patient’s illness. Initial tests and radiologic studies performed on presentation can provide clues that, together with the clinical course and identification of pathogens (Table 92-1), suggest further evaluation or consultation with an immunologist.
Table 92–1 Pathogens Suggestive of Immunodeficiency
| Organism | Disease |
|---|---|
| Pneumocystis jiroveci | HIV, SCID, hyper IgM, XLA |
| Serratia marcescens | Chronic granulomatous disease |
| Aspergillus spp., Nocardia spp. | Chronic granulomatous disease |
| Pseudomonas sepsis | XLA and ARA |
| Mycobacterium/Salmonella spp. | IFN-γ/IL-12 pathway |
HIV, Human immunodeficiency virus; SCID, severe combined immunodeficiency; XLA, X-linked agammaglobulinemia; ARA, autosomal recessive agammaglobulinemia; IFN, interferon; IL, interleukin.
Initial screening laboratory studies for PIDs are outlined in Table 92-2. These include (1) complete blood cell count (CBC), (2) quantitative immunoglobulins, (3) specific responses to vaccinations in immunized individuals, (4) lymphocyte subpopulations, and (5) total hemolytic complement.
Table 92–2 Screening Tests for Evaluation of Immune Function
| WBC and differential | |
| Serum immunoglobulins | IgG, IgA, IgM, IgE |
| Functional antibody responses to immunizations | |
| Lymphocyte subpopulations | T cells: CD3 B cells: CD19 NK cells: CD16/56 |
| Complement studies | Total hemolytic complement |
CBCs can be very informative. Presence or absence of leukocytosis may suggest a normal or abnormal response of the marrow to infection. Cytopenia such as neutropenia (as indicated by the absolute neutrophil count) is a common feature of a series of PIDs and may allow their recognition in context of other findings.6 Total lymphocyte count should be calculated and matched to appropriate aged-matched controls.3 It is important to recognize that the absolute lymphocyte count (ALC) is highest during the first year of life.7 An ALC of less than 2000 cells/μL is not a common consistent finding in consecutive CBCs in infants younger than 1 year. This finding warrants further investigation because it may be the first objective data to suggest a diagnosis of combined immunodeficiency.8 Anemia may indicate autoimmunity and in this context is characteristic of several defects of immune function.2 Thrombocytopenia in a child with severe eczema should prompt the clinician to review the peripheral smear carefully with a pathologist to determine the platelet size; this would allow the diagnosis of Wiskott-Aldrich syndrome. The careful examination of the peripheral smear can identify presence of Howell-Jolly bodies, which are seen in asplenia. Unique morphologic abnormalities of neutrophils are pathognomonic of diseases such as Chediak-Higashi syndrome (Figure 92-2).
Quantification of immunoglobulin (Ig) G, IgA, IgM, and IgE should be performed. As is the case with ALCs, the levels of immunoglobulins in circulation is influenced by the age of the child.3 At birth, term infants have serum IgG levels that are equal to the maternal level or exceed it by 5% to 10%.9 Transplacentally acquired antibodies disappear rapidly after birth, reaching a nadir at 3 months. Average concentrations at this time are 60 mg/dL for infants born at 25 to 28 weeks of gestation, 104 mg/dL for infants born at 29 to 32 weeks, and 430 mg/dL for infants born at term.3,10,11 However, these low levels of maternal IgG antibody appear to be associated with less risk of infection than might be expected. A longitudinal study of the ability of preterm infants to form specific antibodies provides a partial explanation for this low risk.12 By about 9 months of age, infants have formed specific IgG antibodies in response to immunization with diphtheria, tetanus toxoids, and pertussis vaccine. Also by about this age, their antibodies have functional opsonic activity against Escherichia coli and coagulase-negative staphylococci. The ability of newborn infants to produce an active antibody response to antigenic stimulation develops in an orderly fashion. An adult pattern of antibody responses is not acquired until 4 to 5 years of age. Analysis of the factors responsible for this developmental pattern is complex because production of antibody depends not only on B lymphocyte maturity, but also on interactions with other cells that may mature at different rates.
Fetuses can produce serum antibodies and IgM, predominantly in response to intrauterine infection.13 Preterm infants respond nearly as well as term infants to immunization beginning at 2 months with DTP, poliovirus, and hepatitis B vaccines.14–16 Term infants immunized or infected during the first few days of life usually produce protective antibody responses, although at somewhat lower levels than do adults.17–19 The presence of fetal bone marrow B lymphocyte pools similar in size to those of adults and with comparable isotype diversity suggests that their functional deficits may reflect a developing memory repertoire with increased specificity to antigen or the need to generate T lymphocytes capable of producing strong B lymphocyte signals, rather than inherent B lymphocyte immaturity as a sole factor.
Responses to childhood immunizations are very helpful for evaluation of the functional antibody responses. Appropriate responses speak to cognate T- and B-cell interaction necessary for T-dependent antigen responses, and polysaccharide responses speak to antigen responses that do not require T cell help, such as those of the blood group antigens. The collection of 1 to 3 mL of serum that is saved before any gamma-globulin administration is a useful practical recommendation because subsequent serology studies will be influenced by the specific gamma-globulin content of the unit delivered to the patient.
Analysis of lymphocyte subpopulations by flow cytometry is essential when defects of adaptive immunity are entertained. Circulating lymphocytes are distinguished by unique surface molecules that permit their identification. Cluster of differentiation (CD) is a surface marker that identifies a particular differentiation lineage recognized by a group of monoclonal antibodies. Classification of lymphocytes by CD antigen expression is widely used in clinical medicine. Thus T cells are identified as carrying CD3 helper T cells; CD3 and CD4 cytotoxic T cells; CD3 and CD8 NK cells by CD16/56. CD19 identifies circulating B cells. As with ALC, the percent and absolute numbers of different circulating lymphocytes is age dependent.7 In general terms, two thirds of circulating lymphocytes are T cells and one third are B cells; two thirds of T cells are CD4+ and one third are CD8+.
These initial screening laboratory tests will rule out the following PIDs (see Table 92-2): (1) antibody defects such as X-linked agammaglobulinemia (XLA), IgA deficiency, common variable immunodeficiency (CVID), or transient hypogammaglobulinemia of infancy (THI); (2) cellular defects such as severe combined immunodeficiency (SCID), DiGeorge syndrome, and Wiskott-Aldrich syndrome; (3) phagocytic defects defined by neutropenia; and (4) defects in the classic complement pathway.
Clinical Presentations of Congenital Immunodeficiency Syndromes
Combined T- and B-Lymphocyte Defects
Severe Combined Immunodeficiency
Severe combined immunodeficiency (SCID) was first recognized in the late 1950s and was subsequently divided into Swiss-type agammaglobulinemia (i.e., autosomal recessive) and sex-linked lymphopenic immunologic deficiency.20 X-linked SCID (XLSCID), today known as common gamma-chain deficient SCID or γc-SCID, is the most common of all forms of SCID. This combined immunodeficiency is due to mutations in the common γ-chain, an essential signaling molecule. XLSCID comprises 40% to 50% of all SCID phenotypes. Five different cytokine receptors (for IL-2, -7, -9, -15, and -21) require signaling through this molecule. Fourteen different autosomal recessive forms of SCID have been identified.2 The common characteristic of all forms of SCID is the occurrence of a block in the differentiation of T cells, whereas B cells may or may not be present, but are unable to function, and normal or abnormal development of NK cells occurs. Cytokine-dependent signaling, premature lymphocyte cell death, defective recombination or assembly of the antigen receptor, and inability of the T cells to egress effectively from the thymus explain some of the molecular mechanisms that result in the different SCID phenotypes (Table 92-3).2 The incidence ranges from 0.89 to 1.40 per 100,000 live births/year21–23; these disorders are fatal if not promptly recognized and treated.
Table 92–3 Lymphocyte Phenotype Pattern and Genotype in SCID
| Lymphocyte Phenotype | Main Mechanism | Molecular Defect |
|---|---|---|
| T−B+NK− | Signaling pathway | γC, JAK3 |
| T−B−NK− | Cell death | ADA, PNP |
| T−B−NK+ | Ag receptor assembly | RAG1/2, artemis, DNA ligase IV, DNAPKcs, cernunos |
| T−B+NK+ | Other signaling molecules | CD45,IL7R, CD3ζ,ε,δ, coronin-1 A |
γC, Common gamma chain; JAK3, Jannus-associated kinase 3; ADA, adenosine deaminase; PNP, purine nucleoside phosphorylase; RAG, recombinase activating gene; DNAPKcs, DNA-activated protein kinase catalytic subunit; IL, interleukin.
Clinical Presentations and Laboratory Findings in SCID
Common clinical presentations among most forms of SCID that should alert the clinician to consider this condition in the differential diagnosis8,24 include (1) normal birth and delivery; (2) age of presentation to medical attention and/or admission to an ICU usually before 1 year of life (frequently after 3 months of life because of the protection provided by transplacental transfer of maternal IgG antibodies); (3) overwhelming infections with common pathogens such as rotavirus, respiratory syncytial virus (RSV), cytomegalovirus (CMV), parainfluenza virus and adenovirus; (4) opportunistic infections such as Pneumocystis jiroveci; (5) persistent oral candidiasis and failure to thrive; (6) chronic hepatitis or sclerosing cholangitis; (7) persistent diarrhea; (8) skin rash that does not resolve with standard therapies; (9) enteroviral encephalitis; and (10) disseminated varicella zoster postvaccination. It is important to recognize that children with SCID, if transfused with nonirradiated packed red blood cells, may develop overwhelming and life-threatening graft-versus-host disease within 1 to 2 weeks because of clonal expansion of donor-derived leukocytes present in the blood product.
Absent thymic shadow on the initial admission chest radiograph and persistent lymphopenia (<2000 cells/μL3) since admission can be easily overlooked. Many cases of SCID could be predicted by reviewing the absolute lymphocyte count at birth.25
Therapy for Patients with Clinical Suspicion or Diagnosis of SCID
While managing a child with suspected SCID, medical therapies should be targeted at both identifying pathogens (often more than one pathogen may be identified in the same individual) and instituting both targeted and prophylactic antimicrobials. Fungal and prophylaxis against Pneumocystis jiroveci should be started. Trimethoprim-sulfamethoxazole (TMP/SMX) may be initiated as early as 1 to 4 weeks of age with close monitoring of liver function and transaminases.26 Storage of 1 to 3 mL of serum before gamma-globulin infusions is helpful, as previously mentioned. Gamma-globulin should be give at doses of 400 to 500 mg/kg every 3 to 4 weeks intravenously (IV). If IV access is difficult, subcutaneous administration of IgG is possible. Blood products should be irradiated and CMV negative. No live vaccines such as rotavirus or varicella should be given to the patient or family members in contact with the child. Rotavirus vaccine strains have been reported as causing diarrhea and being the first manifestation in several children with SCID.27
After the diagnosis is made, the patient should be transferred to a center with experience in the management and treatment of these disorders. Human leukocyte antigen (HLA) typing of patient and family and consultation with the stem cell transplant team should be pursued. Without a hematopoietic stem cell transplantation (HSCT), children with SCID die.24 Now considered standard of care, this procedure has achieved the most successful survival record, ranging from 63% to 100% when an HLA-matched sibling donor has been available and 50% to 77% when haploidentical or HLA-mismatched donor is used.28–35 The lack of donor availability, variable evidence of long-term immunologic reconstitution, and other limitations have led to the extensive use of unrelated but matched donors as a source of stem cells for SCID. The outcomes have improved over the recent years, with survivals ranging from 63% to 80%.31–3336
Other Combined Immunodeficiency Disorders
Many other forms of combined immunodeficiency have been described. In general, these are rare conditions, and similar general concepts of diagnosis and management strategies as for SCID should apply. Family histories are helpful because consanguinity is not uncommon. Microcephaly, deafness, ectodermal dysplasia features, cartilage abnormalities, and hematologic abnormalities such as eosinophilia and cytopenia may be clues that can further guide the molecular analysis.2
Hyper Immunoglobulin M Syndrome
Hyper IgM comprises a group of disorders of Ig isotype class switch recombination.37 First described by Rosen et al.,38 it was initially classified as a defect in antibody production. Patients had very low levels of circulating IgG and IgA antibodies and elevated levels of IgM. By 1985, Mayer,39 in critical co-culture experiments, demonstrated that B cells, isolated from hyper IgM patients, could produce IgG and IgA in vitro when incubated with T cells derived from a Sezary T-cell lymphoma and suggested a role for T lymphocytes in Ig isotype switching.39 It was not until 1993 that three independent groups identified CD40 ligand (also known as CD154) as the molecule responsible for the X-linked, and most common form, of hyper IgM (XHIGM).40–42 As molecular mechanisms of class switch recombination have been elucidated, so have the identification of autosomal recessive forms of hyper IgM.43
IgM molecules are not opsonins because there are no receptors on phagocytes for the Fc portion of this immunoglobulin molecule. Phagocytosis is promoted by complement activation, C3 deposition on the bacterial surface, and subsequent phagocytosis. XLHM patients are deficient in antibody responses that require T-cell help. Examples include protein antigens such as bacterial toxins or bacterial capsules wall proteins as seen in Staphylococcus pyogenes. The “combined” nature of this immunodeficiency is due to the role that CD40 ligand plays in the effector function of both macrophages and T cells. For example, activation of macrophages against opportunistic infections such as Pneumocystis jiroveci requires binding of CD40 found on the macrophage surface to its ligand, which is expressed on an activated T cell. This T cell/macrophage interaction allows for activation of the macrophage and subsequent microbial killing by intracellular production of oxygen radicals, nitric oxide, and lysosomal enzymes. Therefore these patients are susceptible to infections similar to those seen in other hypogammaglobulinemic states and pathogens that require intact T-cell function such as viruses, fungi, and parasites.44–46
Clinical and Laboratory Presentation of X-Linked Hyper Immunoglobulin M
This is a rare immunodeficiency that becomes manifest in most cases before the age of 4 years. In a cohort of 79 patients studied, Pneumocystis pneumonia was the initial presentation in more than 50% of patients.46 Neutropenias (seen in >50% of cases), other cytopenias, sclerosing cholangitis–associated cryptosporidium infection, hepatitis, progressive neurodegeneration,47 and gastrointestinal malignancies48 contribute to morbidity and mortality.45,46 When a patient presents to the ICU in respiratory failure and P. jiroveci is isolated from bronchoalveolar lavage, after HIV is ruled out three PIDs should come to mind: SCID, hyper IgM, and X-linked agammaglobulinemia (XLA).
In contrast, patients with a rare autosomal recessive form of hyper IgM due to mutations in activation-induced cytidine deaminase (AID), an enzyme involved in isotype switching, come to medical attention in early childhood with recurrent bacterial sinopulmonary and gastrointestinal infections. Opportunistic infections and neutropenia are uncommon. Cellular immunity is normal, and yet massive lymphadenopathy and lymphoid hyperplasia are characteristic.49
Treatment of hyper IgM is directed at replacing antibodies and preventing opportunistic infection. Gamma-globulin infusions at dose of 400 to 500 mg/kg every 3 to 4 weeks, TMP/SMX prophylaxis, and G-CSF as needed for neutropenias are standard therapies. HSCT has also been successfully performed in XHIGM.50






Full access? Get Clinical Tree


